Review
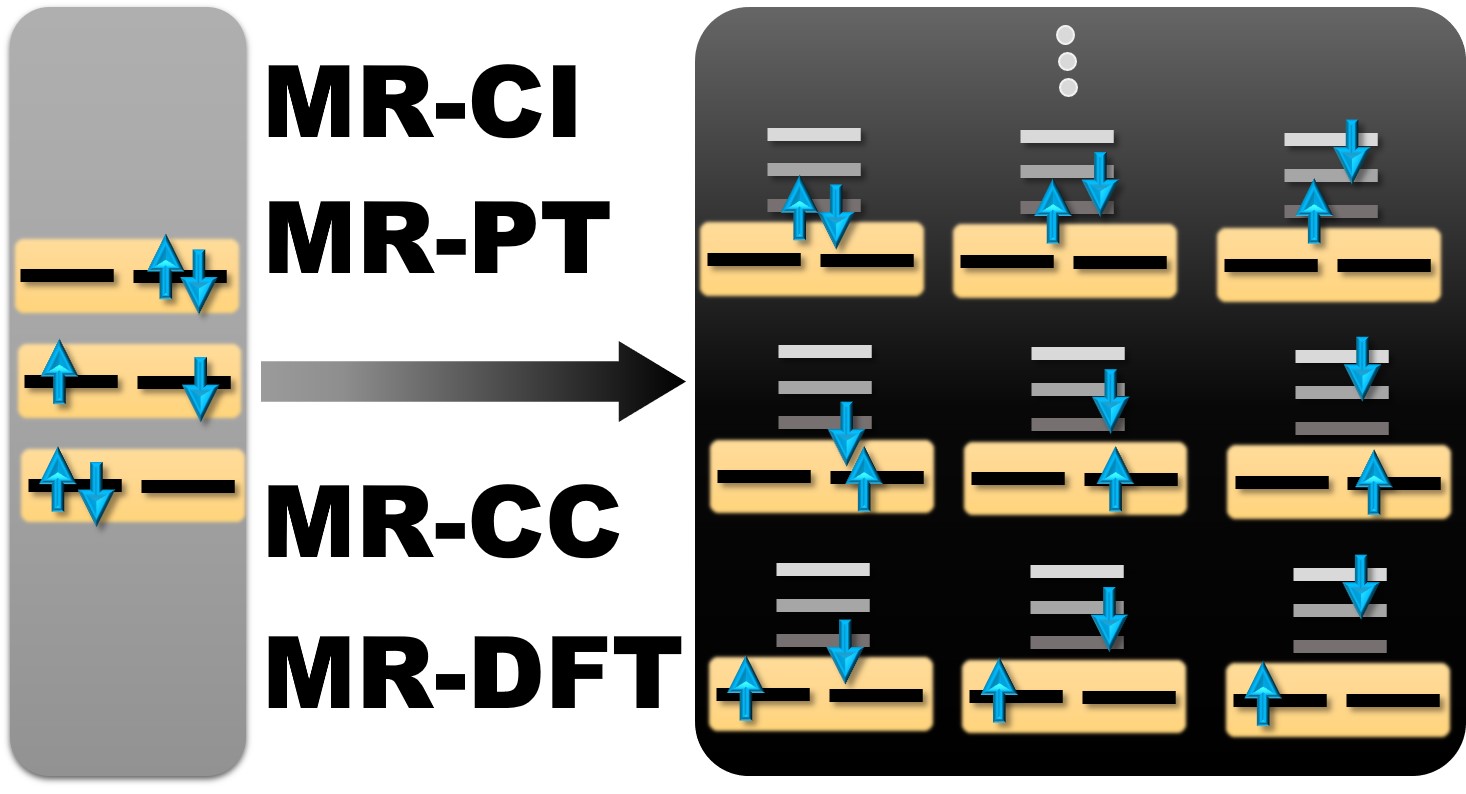
Multireference Approaches for Excited States of Molecules
Paper reviews multireference methods and applications to molecular excited states.
Photosensitization and singlet oxygen
Influence of Sugar on Triplet Decay of Thionucleosides
Functionalization of the sugar group can be used to control the triplet-decay rate of thionucleosides.
Review
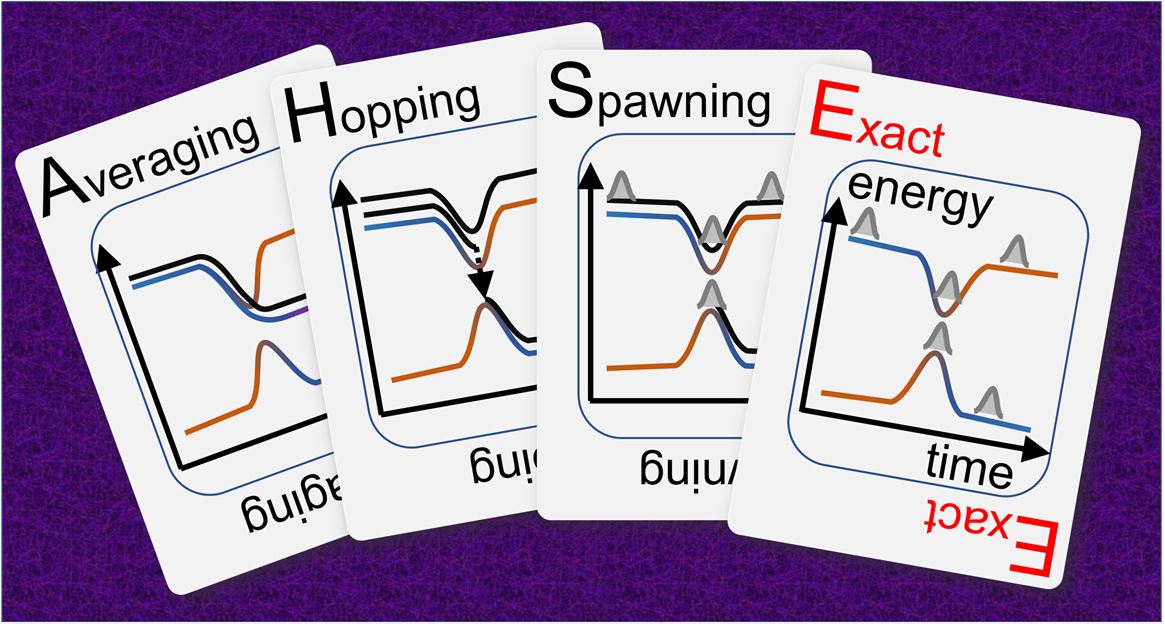
Advances in Nonadiabatic Mixed Quantum-Classical Dynamics
Paper reviews the last ten years of developments in nonadiabatic dynamics.
Methods and Software
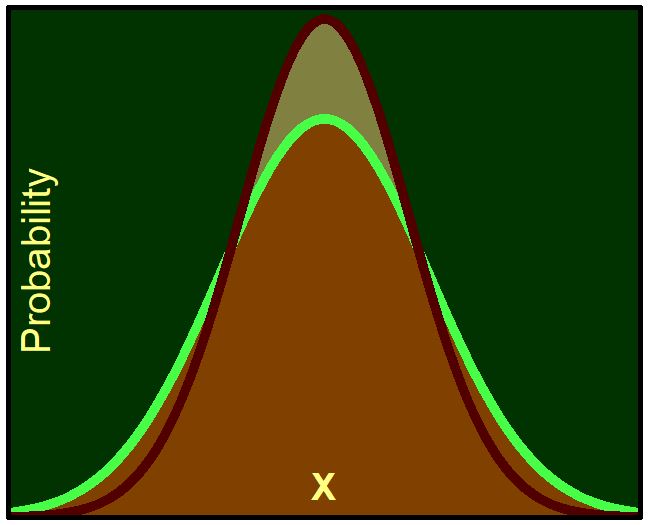
Nuclear Ensemble with Importance Sampling
Mapping between probability distributions can save a lot of time in dynamics and spectrum simulations.

Quantum chemistry
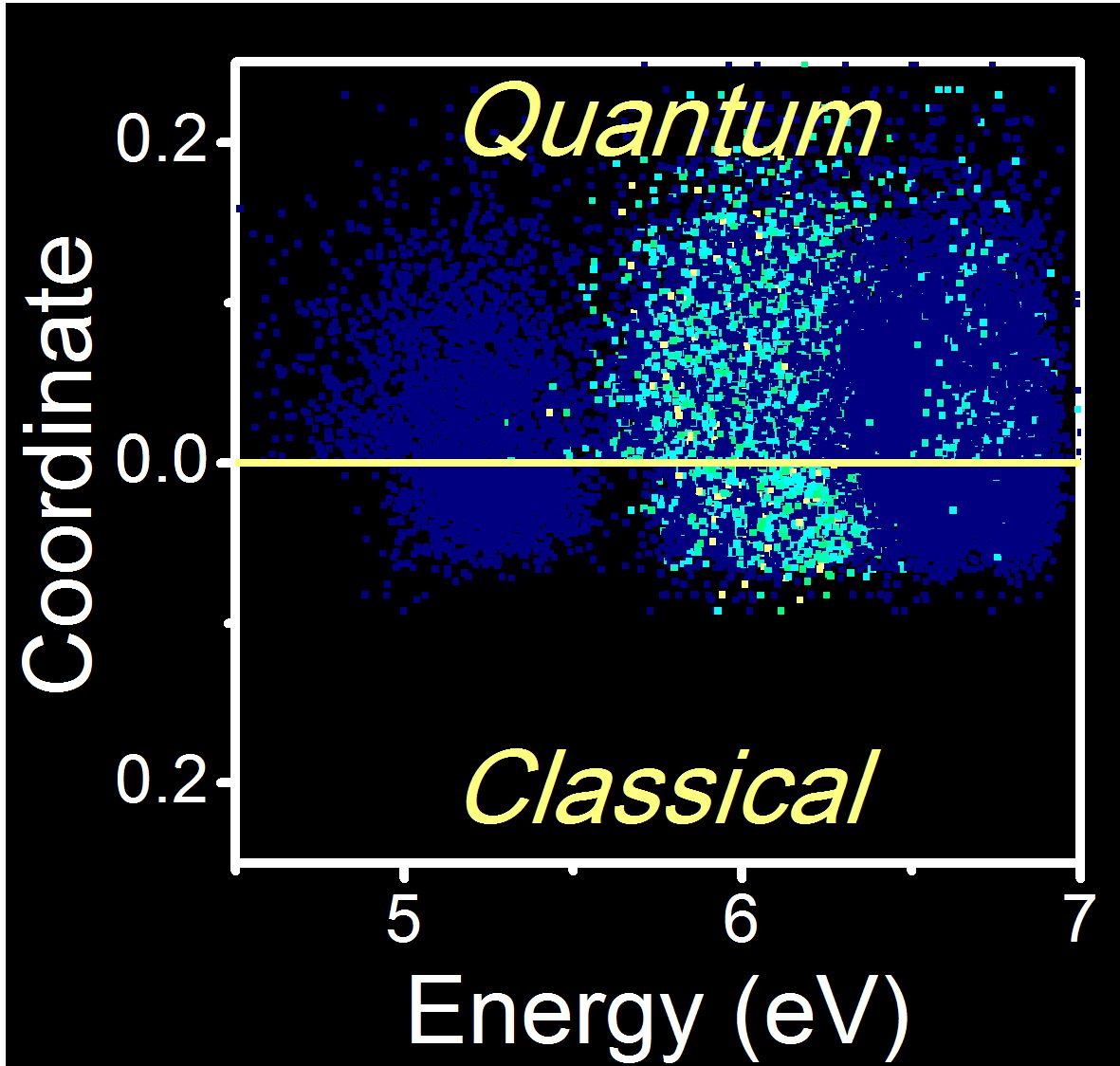
How Many Trajectories Should I Run?
Want to do dynamics? This is how many trajectories you will need. (more…)
Methods and Software
Surface Hopping with TD-DFTB
Interface between Newton-X, DFTB+, and TheoDORE enables nonadiabatic dynamics at nanoscale.
Photosensitization and singlet oxygen
Singlet Oxygen: Rates Strongly Depend on Geometry
Simulations show how to maximize reaction rates for singlet oxygen generation.
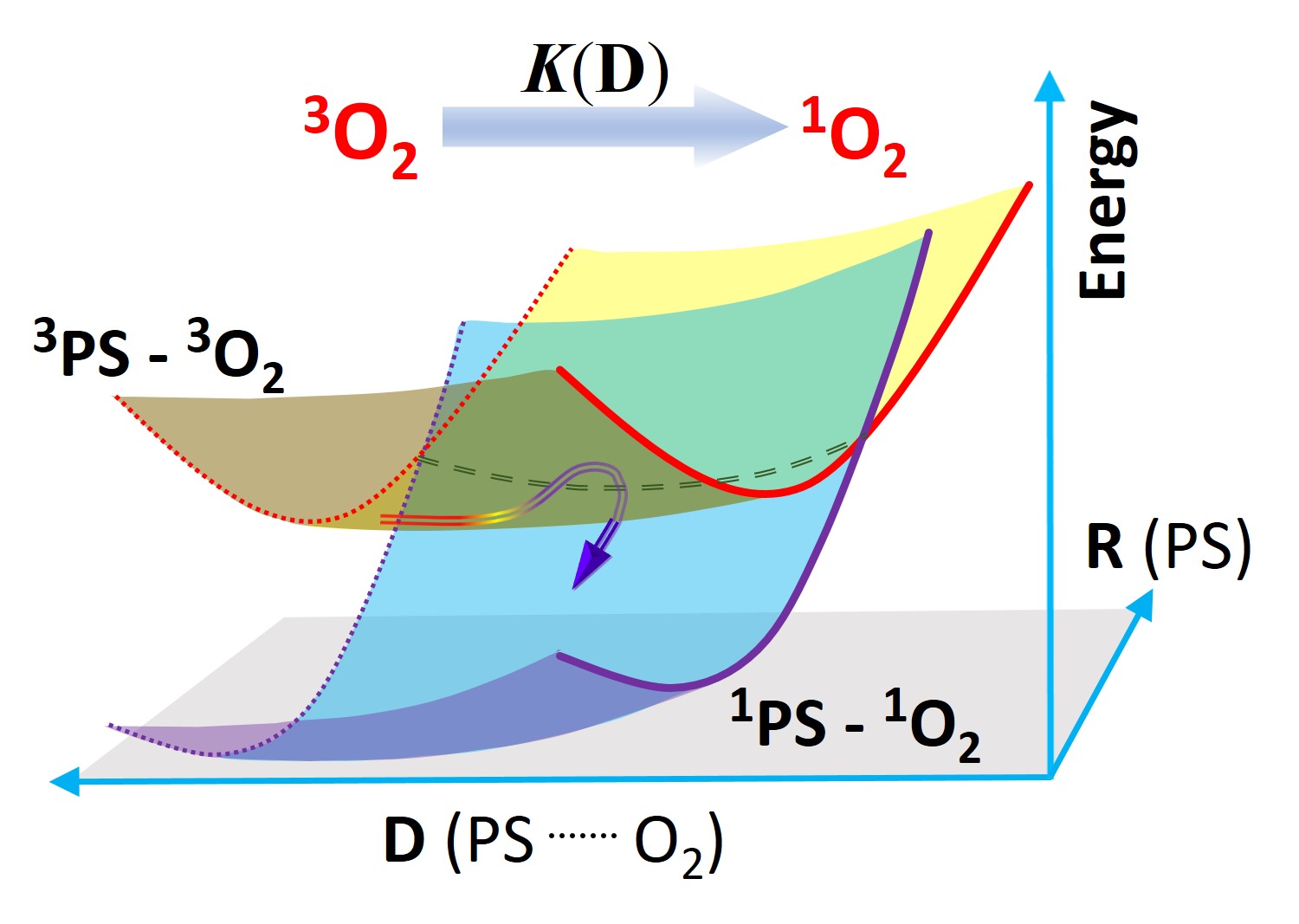
Methods and Software
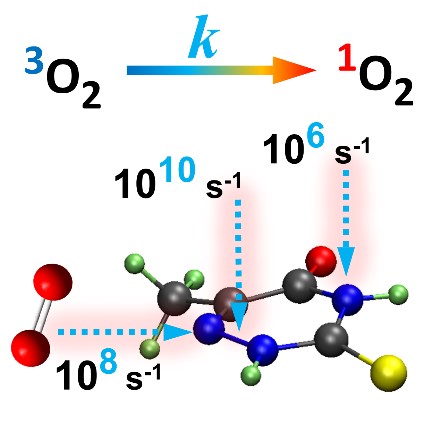
A Kinetic Model for Singlet Oxygen Photogeneration
Reaction rates for energy transfer calculated from first principles.
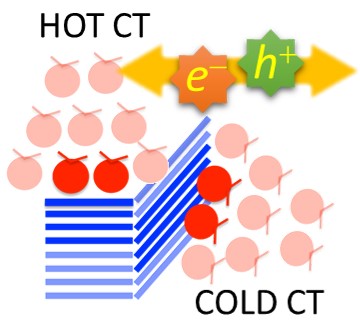
Organic photodevices
Hot and Cold Charge Transfer in Organic Donor/Acceptor Interfaces
Charge separation follows different photophysics in different donor/acceptor arrangements.
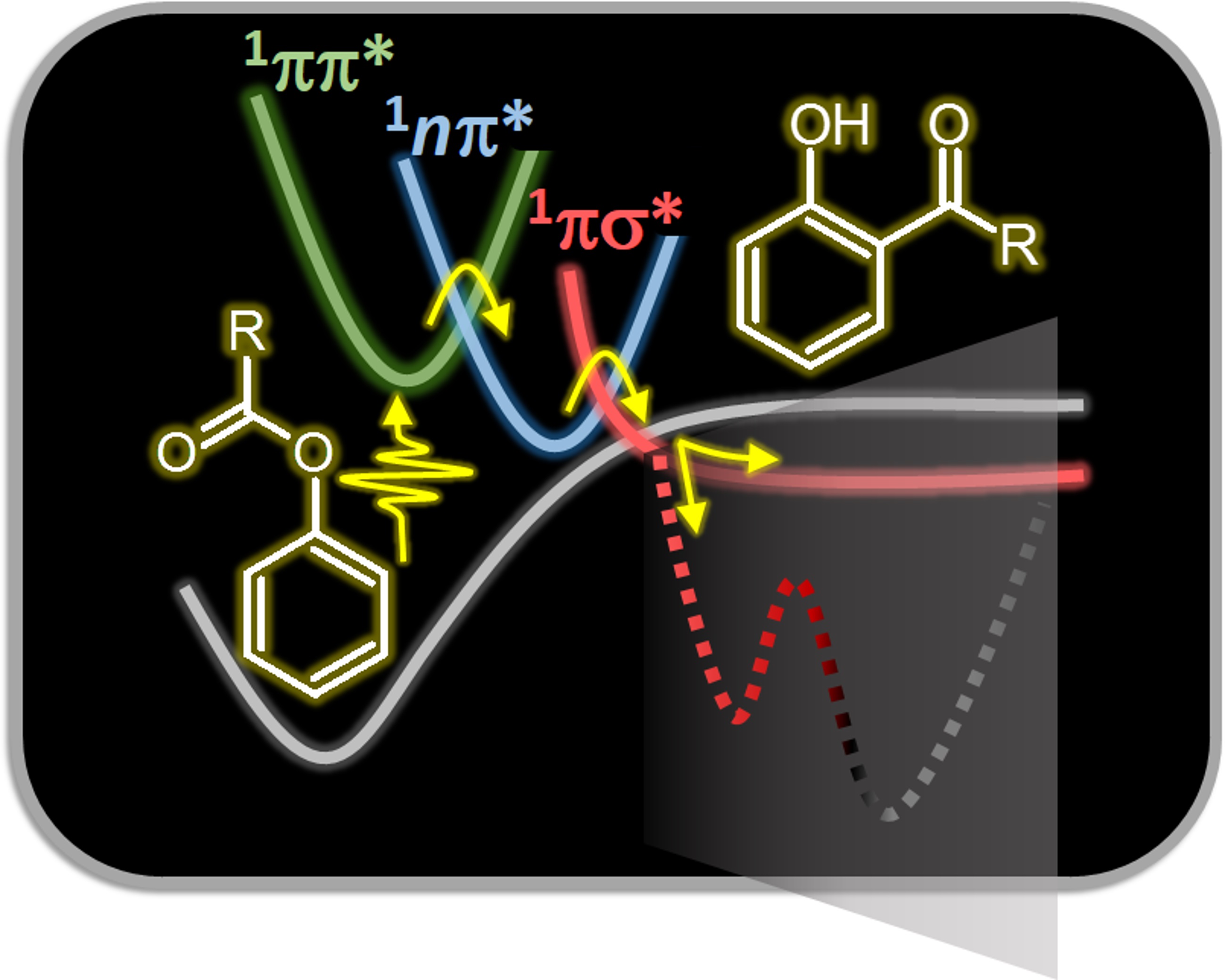
Photochemistry and photophysics
Three-state Model for the Photo-Fries Rearrangement
Photo-Fries rearrangement plays a central role in organic synthesis. But how does it really work?
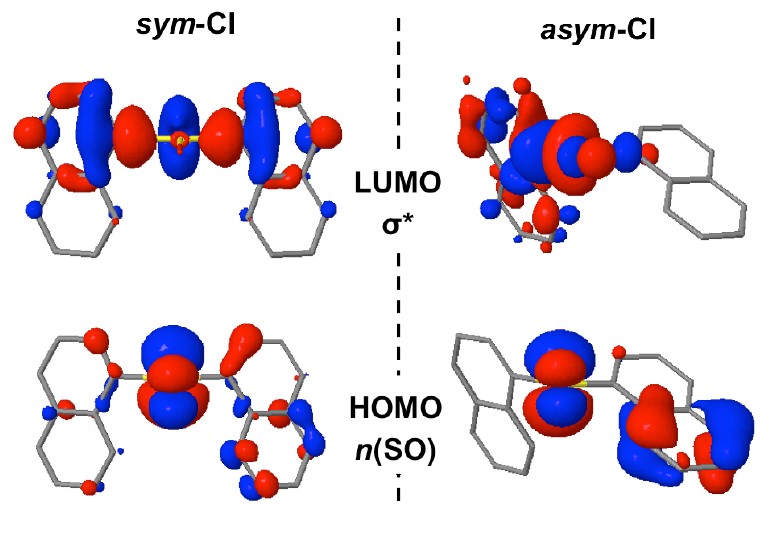
Organic photodevices
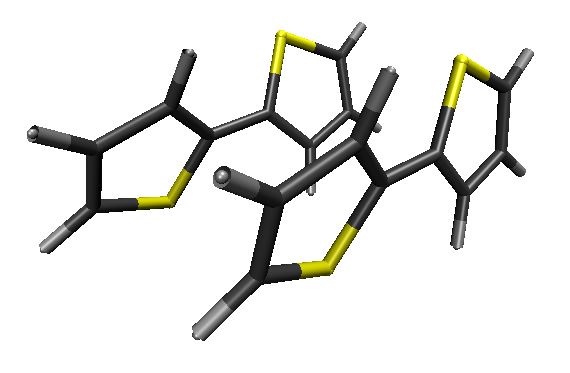
Photophysics of Sulfur-Bridged Naphthalene Dimers
Sulfur bridge’s oxidation state dramatically changes the photoluminescence of bichromophores. That’s why.
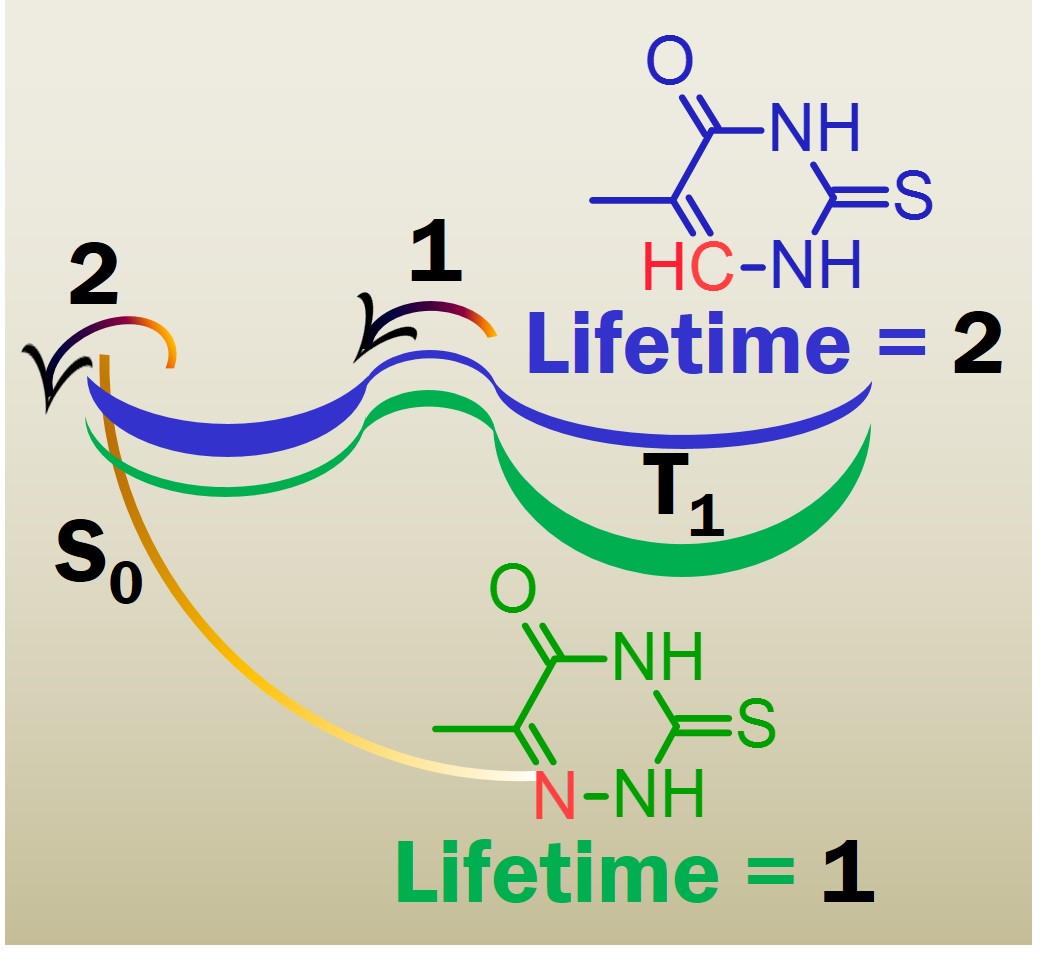
Photosensitization and singlet oxygen
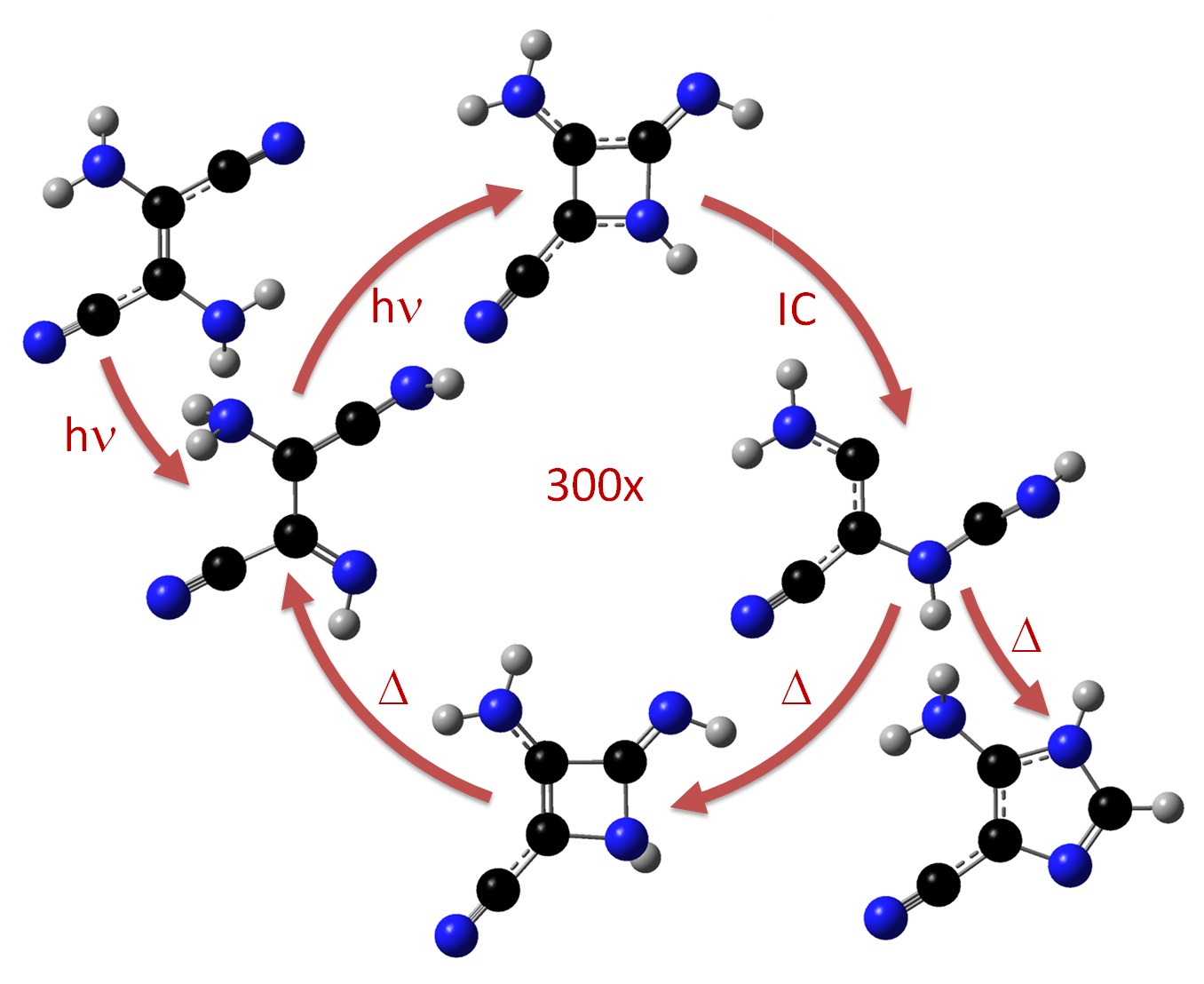
On the Decay of the Triplet State of Thionucleobases
Double-well shaped triplet state in thionucleobases can be used to tune singlet oxygen yields.

Quantum chemistry
What’s the Biggest System We Can Do Dynamics?
Want to do dynamics? This is how much it’ll cost.
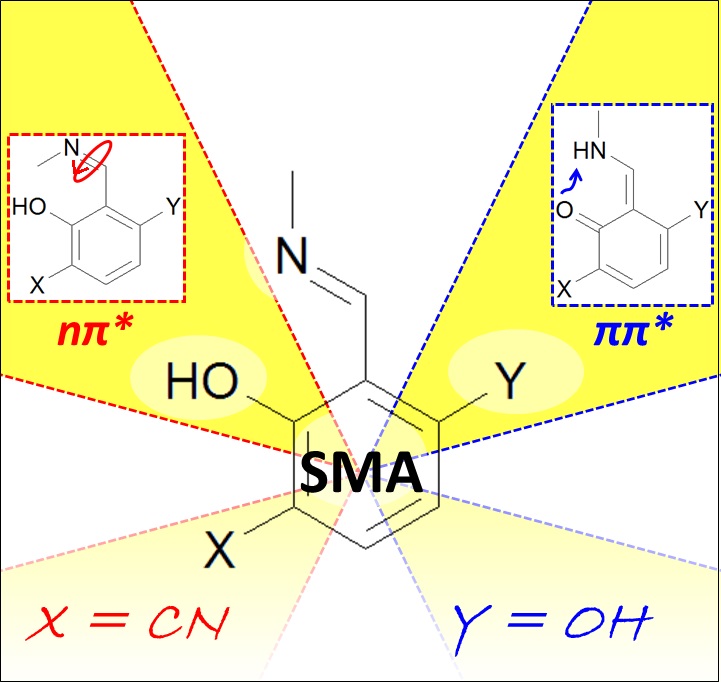
Photochemistry and photophysics
Tailoring Schiff Bases Photoswitching
Dynamics reveals how to design chemical substitutions to control proton transfer efficiency.
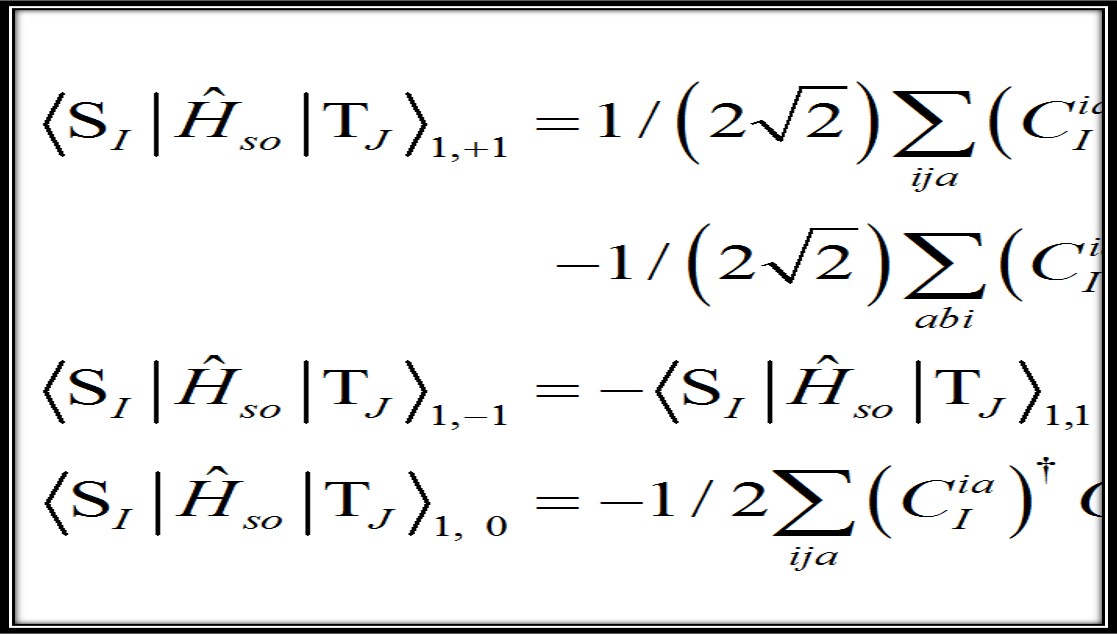
Methods and Software
Spin-Orbit Couplings Based on Density Functional Theory
PySOC, a new program for fast and flexible computation of spin-orbit couplings.
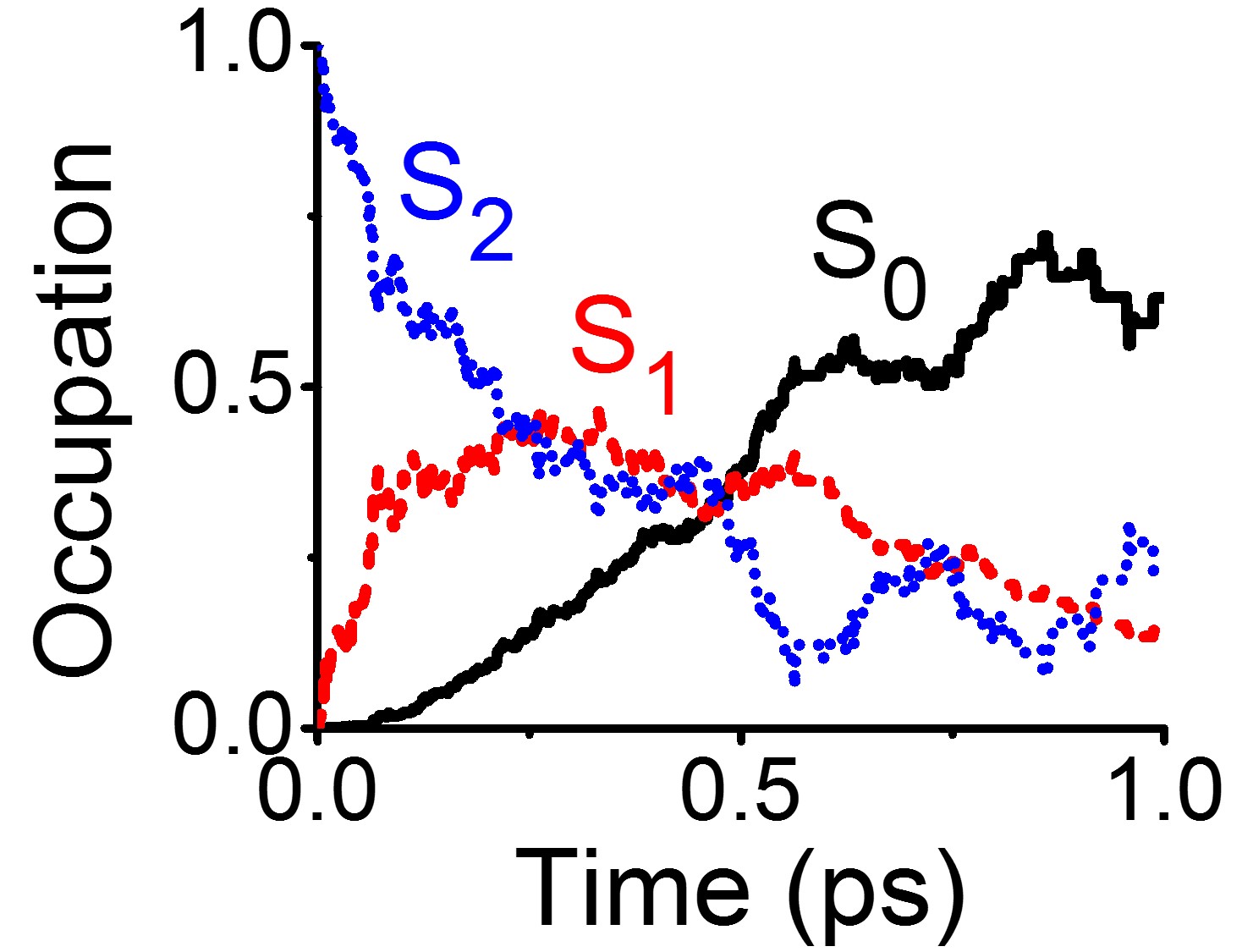
Photochemistry and photophysics
Revisiting the Excited State Dynamics of Thymine
So far, the long lifetime of UV-excited thymine has been explained in terms of trapping in the second excited state. Simulations with electron correlation shows this hypothesis doesn’t hold.
Atmospheric photochemistry Quantum chemistry
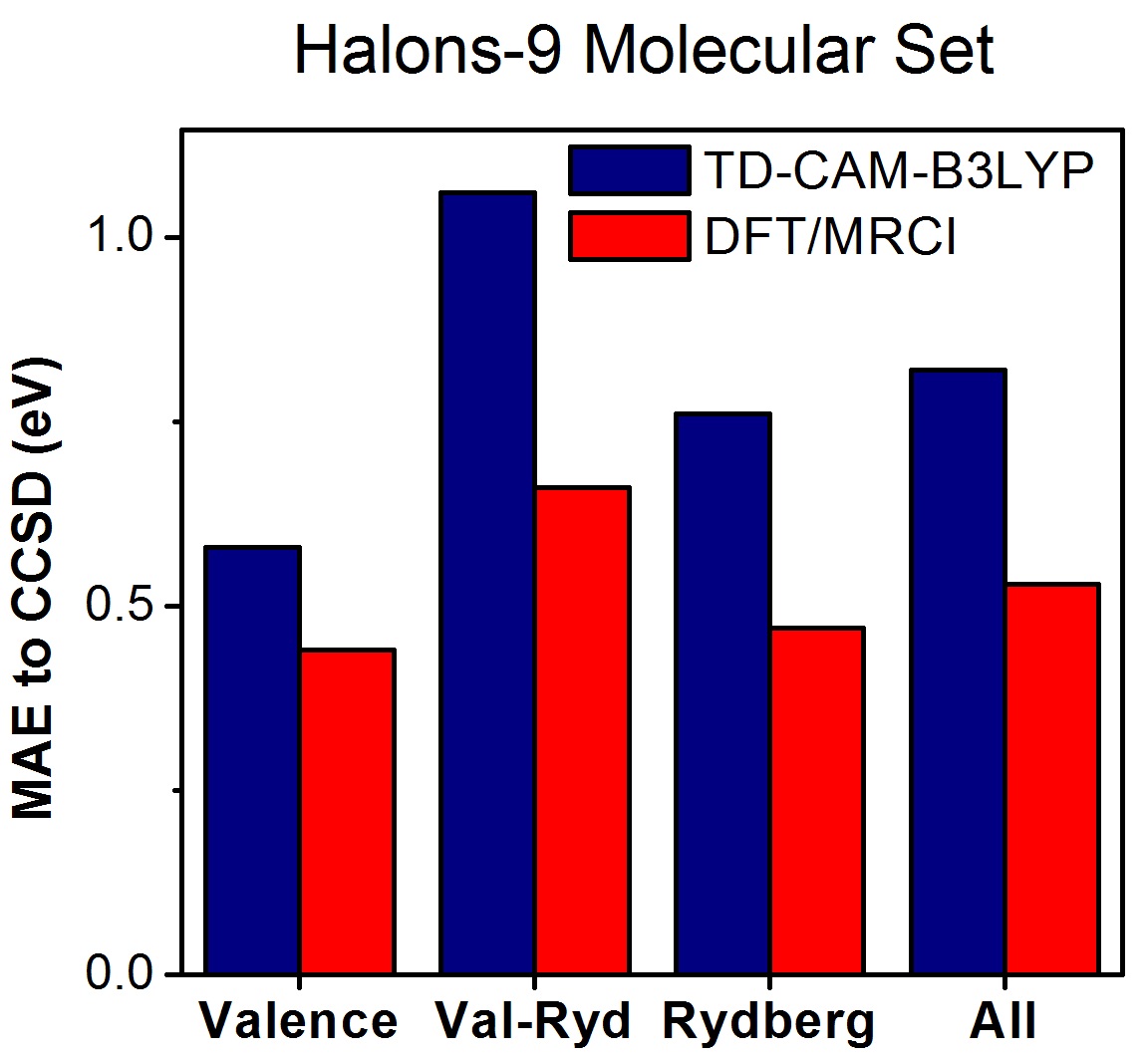
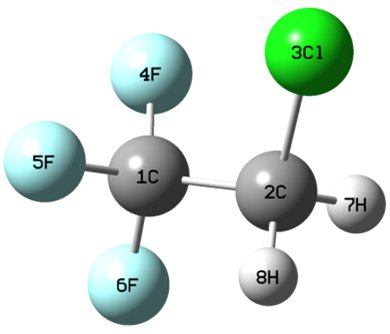
The Halons-9 Molecular Set
A new molecular set composed of small haloorganic compounds shows that popular computational methods may have errors of about 0.8 eV.
Methods and Software
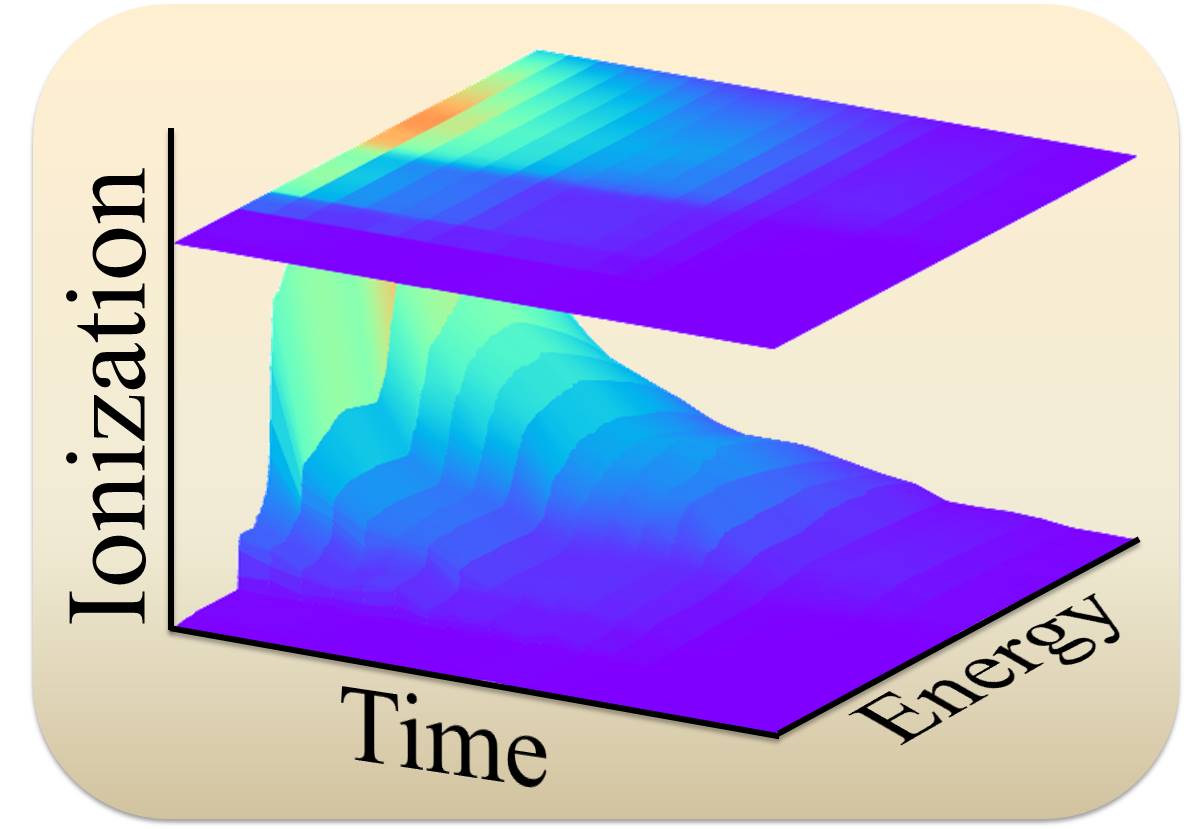
Photoelectron Spectrum Simulations
Methods based on nuclear ensembles allow to simulate steady and time-resolved photoelectron spectra with absolute intensities.
Photochemistry and photophysics
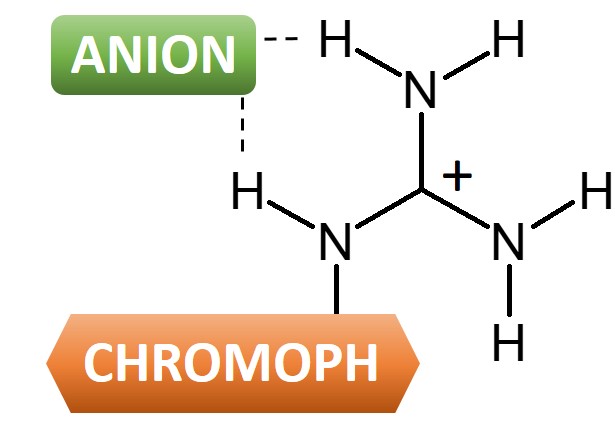
Guanidinium: A New Analytical Tool to Detect Anions
Chromophore-guanidine compounds can be used as selective anion sensors.
Photosensitization and singlet oxygen
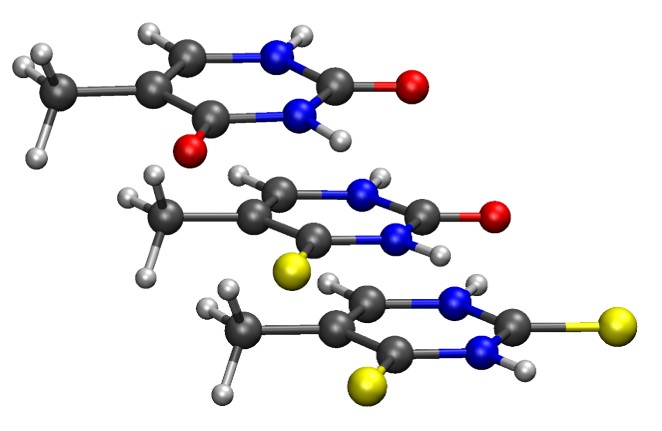
Explaining the Photophysics of Thiothymines
Why does replacing different oxygens of thymine with sulfur cause distinct absorption and intersystem crossing?
Organic photodevices
Organic Photovoltaics: Make Light, Not Heat
A new approach based on dynamics simulations is proposed to boost the efficiency of organic photovoltaics.
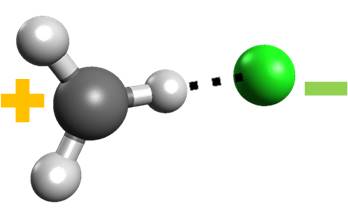
Atmospheric photochemistry Quantum chemistry
Chlorine Hydrogen Bonds? Yes, They do Exist
CH∙∙∙Cl hydrogen bond in chloromethane may be formed by UV irradiation.
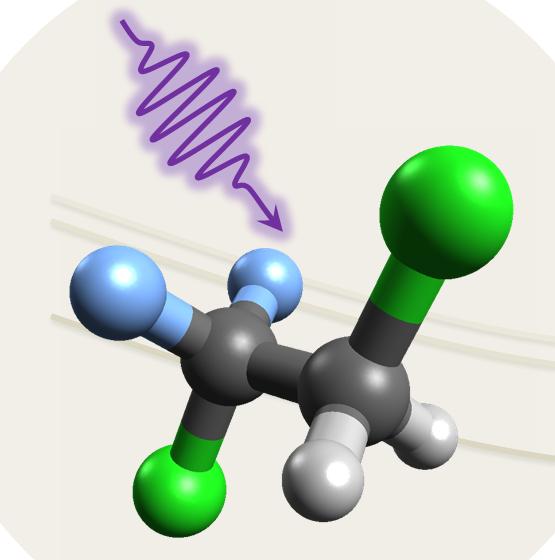
Atmospheric photochemistry
Nonadiabatic Dynamics of HCFC-132b
HCFC-132b is an important industrial compound with strong environmental impact. Nonadiabatic dynamics explains how UV induces its photo-decomposition in the sub-picosecond scale.
Methods and Software
Sampling Initial Conditions for Dynamics and Spectrum
Wigner distribution or ground-state trajectories, what is the best way to sample initial conditions for excited-state dynamics? (more…)
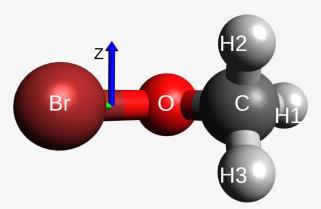
Atmospheric photochemistry
Photochemistry of Methyl Hypobromite
Excited states of bromites are true challenges for theoretical chemistry. It’s time to face them. (more…)
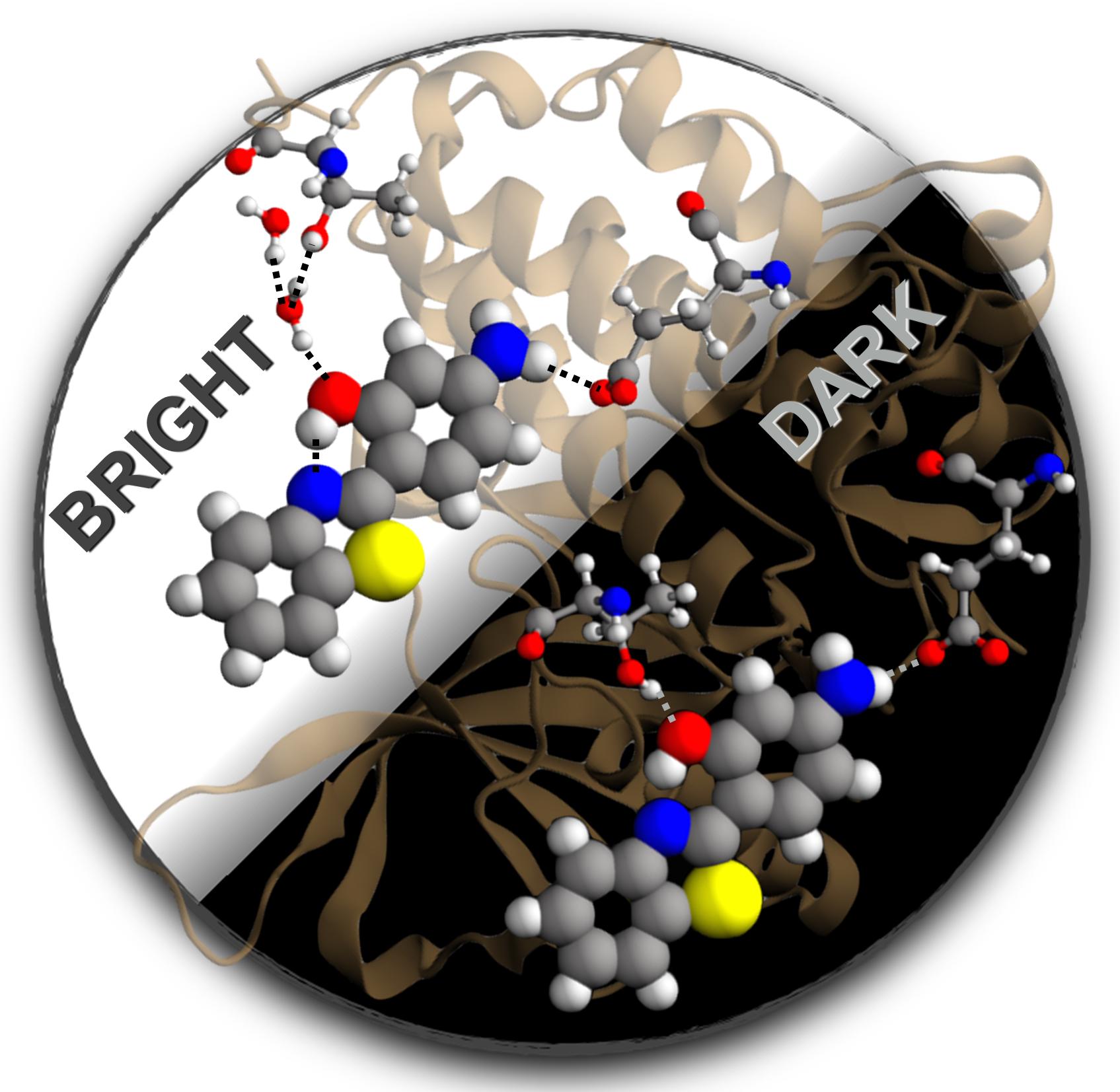
Excited state proton transfer
Excited-State Proton Transfer Can Tune the Color of Protein Fluorescent Markers
Excited-state proton transfer may give rise to new diagnostic tools to follow the clinical evolution of cancer patients.
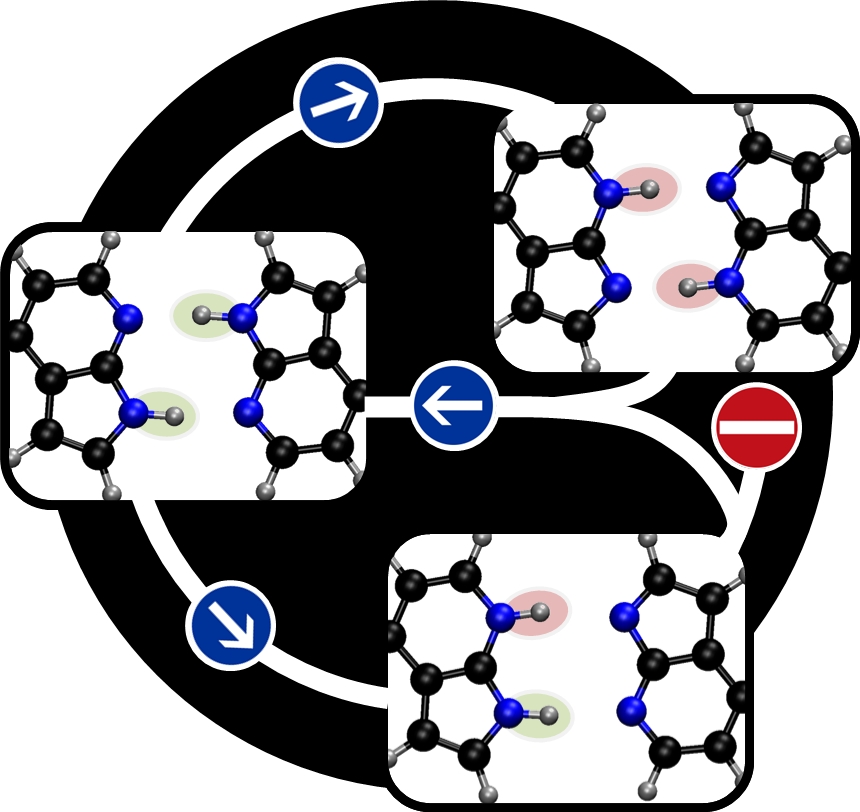
Excited state proton transfer Quantum chemistry
Stepwise Proton Transfer Is Not Possible in 7-Azaindole Dimer
Topographical analysis of the dimer’s excited state shows that internal conversion after first proton transfer blocks the stepwise process.
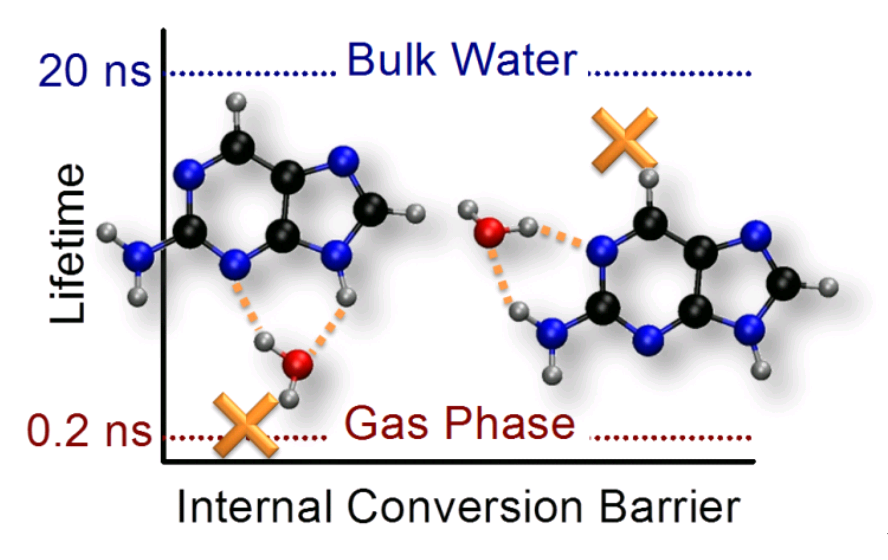
Photochemistry and photophysics
Why One Water Molecule Makes Aminopurine Fluorescent
Using computational simulations of 2-aminopurine-water clusters, we show why a single water may turn the fluorescence of this molecule on.
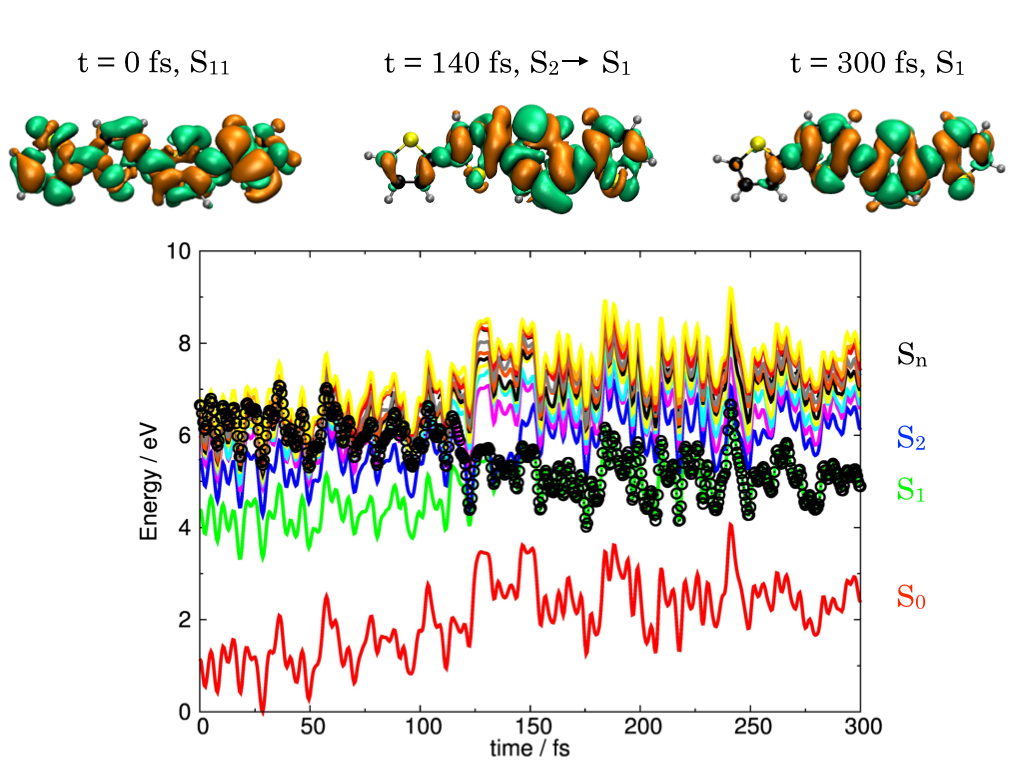
Organic photodevices
Exciton Relaxation in Oligothiophenes
Ultrafast dynamics simulations show how excitons localize in an organic polymer.
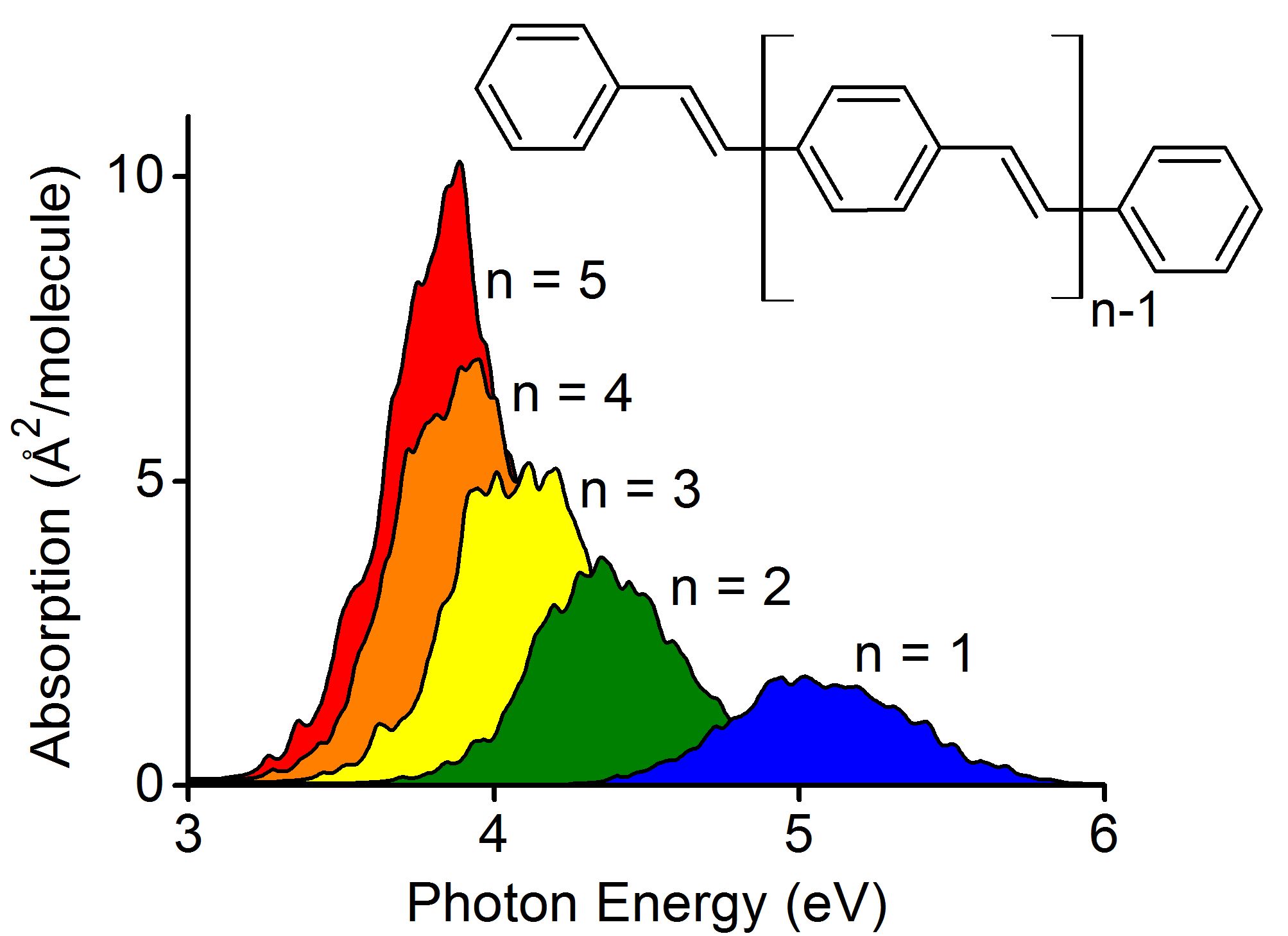
Photochemistry and photophysics
Absorption and Fluorescence Spectra of PPV Oligomers
Computational simulations unveil trends in the absorption and emission spectra of PPVs. (more…)

Review
Photoinduced Processes in Nucleic Acids – Book Chapter
Work reviews what happens to DNA after UV excitations. (more…)
Atmospheric photochemistry
Photochemistry of Hydrochlorofluorocarbons
The photochemistry of an HCFC is explained via ultrafast dynamics simulations. (more…)
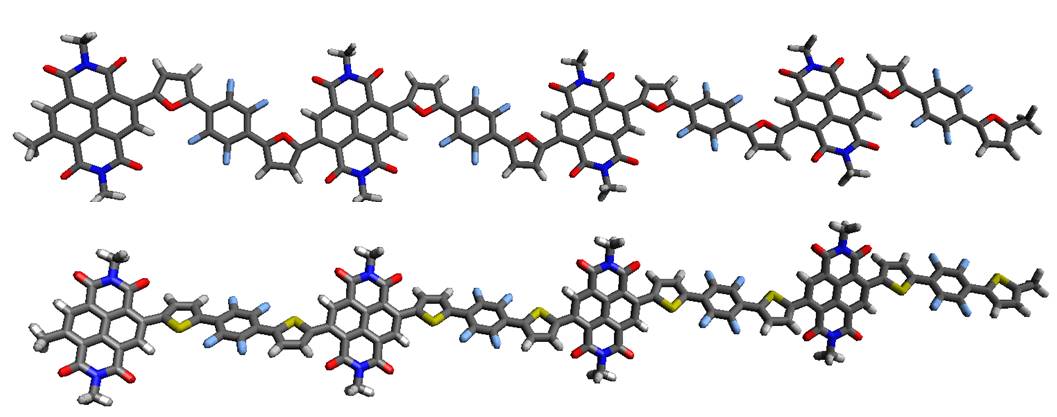
Organic photodevices
High-electron mobility naphthalene diimide copolymers prepared by direct arylation
Synthesis and characterization of organic materials with high electron mobility. (more…)
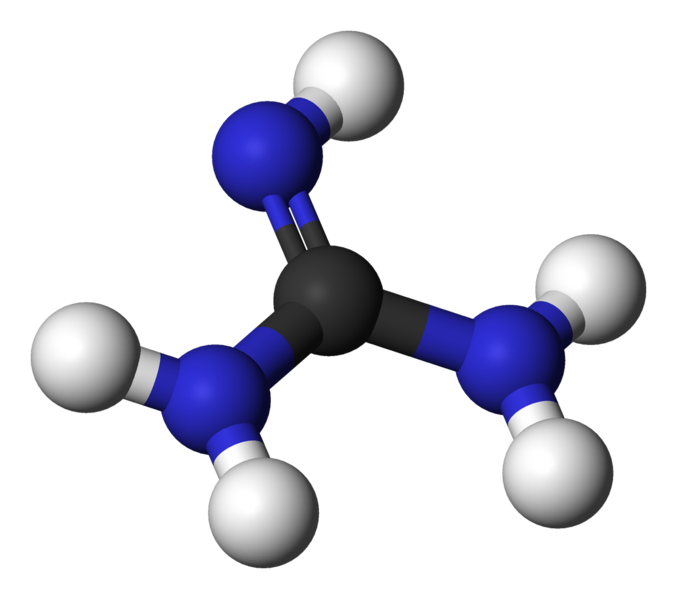
Photochemistry and photophysics
Guanidine and Guanidinium
Paper explains the absorption spectrum of guanidine and guanidinium. (more…)
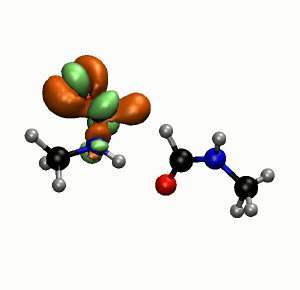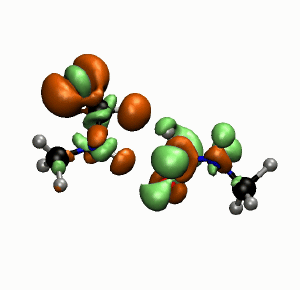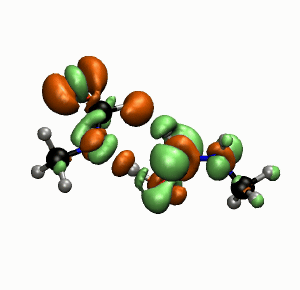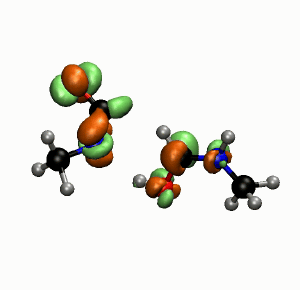
Photobiology
Photostablity of Peptide Bonds
Simulations and experiments show that hydrogen bonds play an important role for the stability of peptide bonds against UV radiation. (more…)
Photobiology
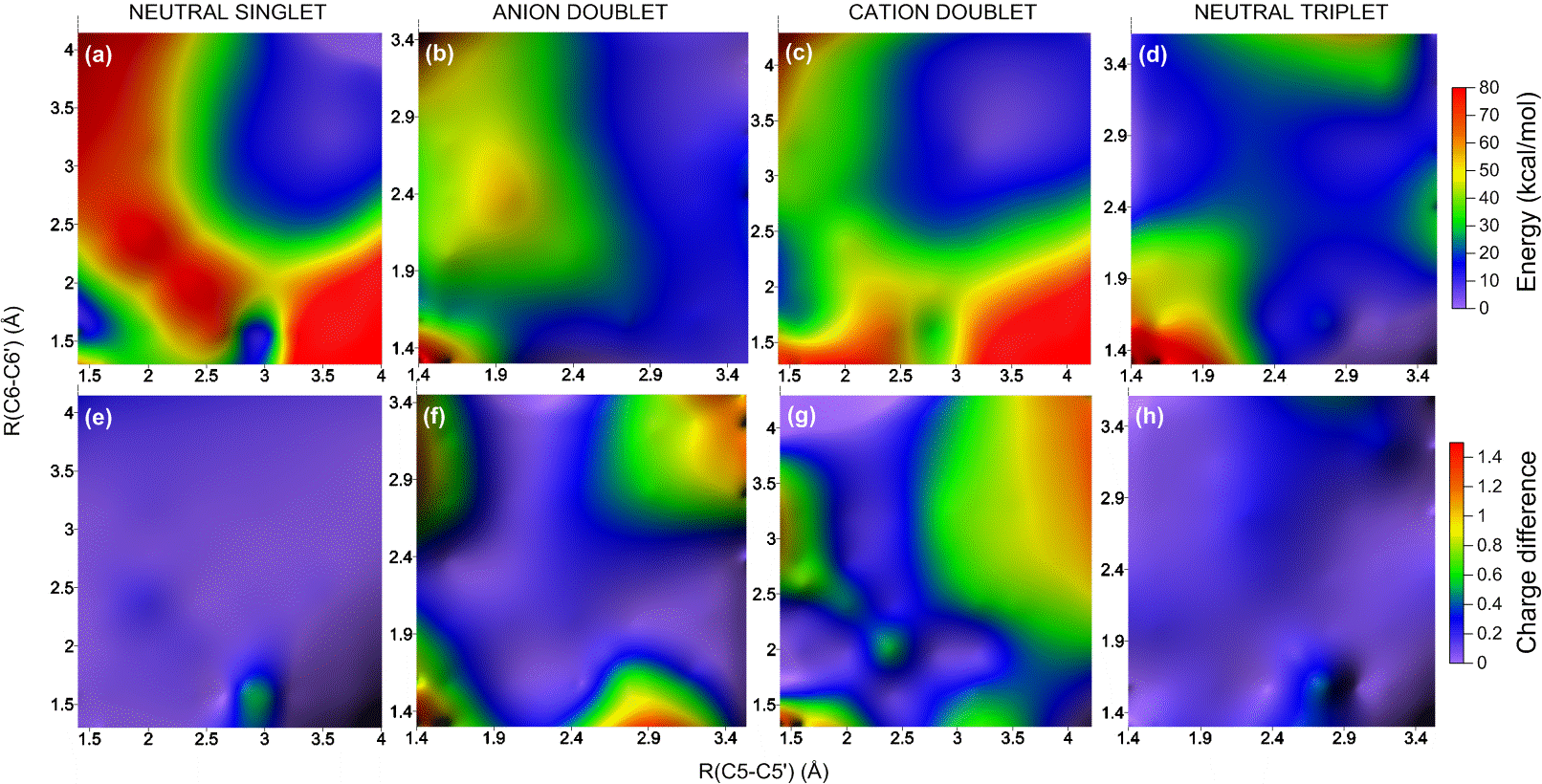
Formation and Repair of Cyclobutane Pyrimidine Dimers
Diverse mechanisms of formation and repair of CPDs with different charge, excitation, and multiplicity states are mapped using high-level ab initio methods. (more…)
Photochemistry and photophysics
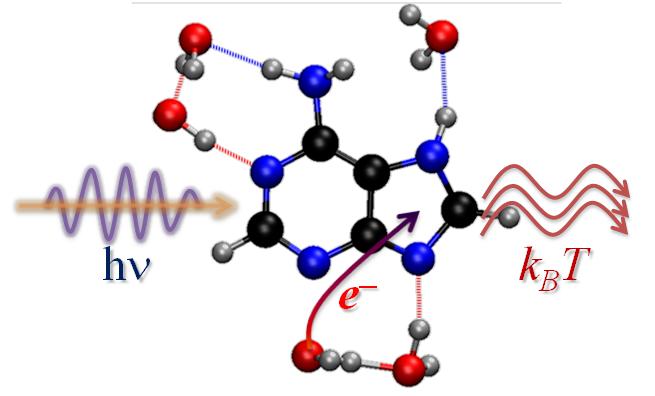
Photorelaxation induced by solvent-chromophore electron transfer
A new internal conversion mechanism induced by electron transfer from solvent to chromophore has been revealed by dynamics simulations. (more…)
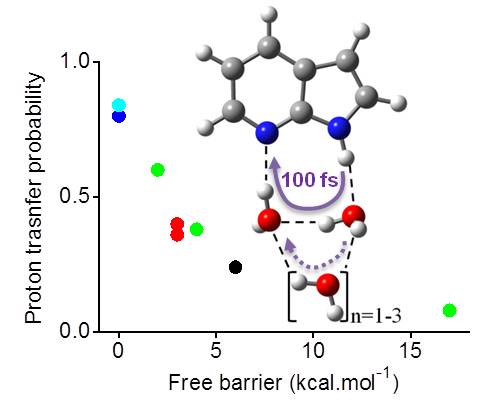
Excited state proton transfer
Excited-state proton transfer in solvated 7-azaindole
Excited state proton transfer in 7-azaindole in water takes place through the first solvation shell. (more…)
Methods and Software
Dynamics with MCSCF
An interface between GAMESS and Newton-X is available for surface hopping simulations. (more…)
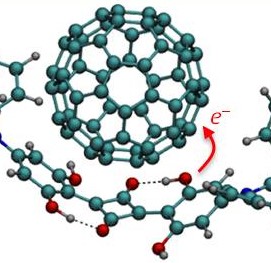
Organic photodevices
Squaraine-Fullerene Organic Interfaces
Squaraine-fullerene complexes show a non-Marcus regime. (more…)
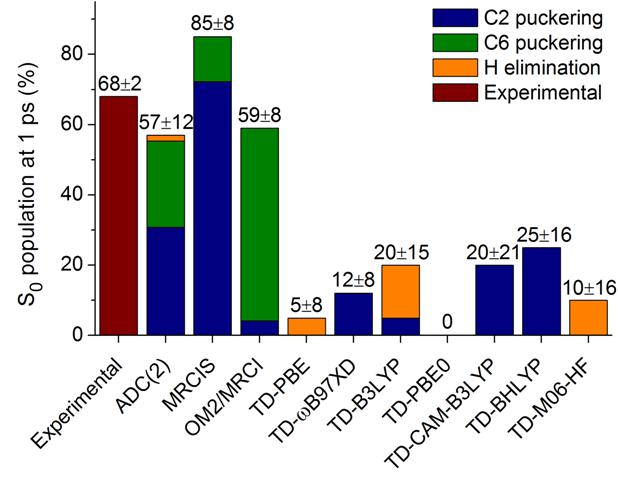
Photobiology
Benchmark of Dynamics Simulations: Adenine
Benchmark of excited-state nonadiabatic dynamics simulations for adenine shows the limits of quantum chemical methods. (more…)