When and how to do surface hopping with DFT.
The demands in the field of nonadiabatic dynamics are quickly growing, and the development of surface hopping based on density functional theory (SH/DFT) has been a major advance.
In a recent review to Topics in Current Chemistry, Rachel Crespo-Otero and I have discussed in details the surface hopping approach, the methods for computation of excited states based on DFT (not only TDDFT!), the connection between these methodologies, and their diverse implementations.
The shortcomings of the methods are critically addressed and a number of case studies from diverse fields are surveyed as well.
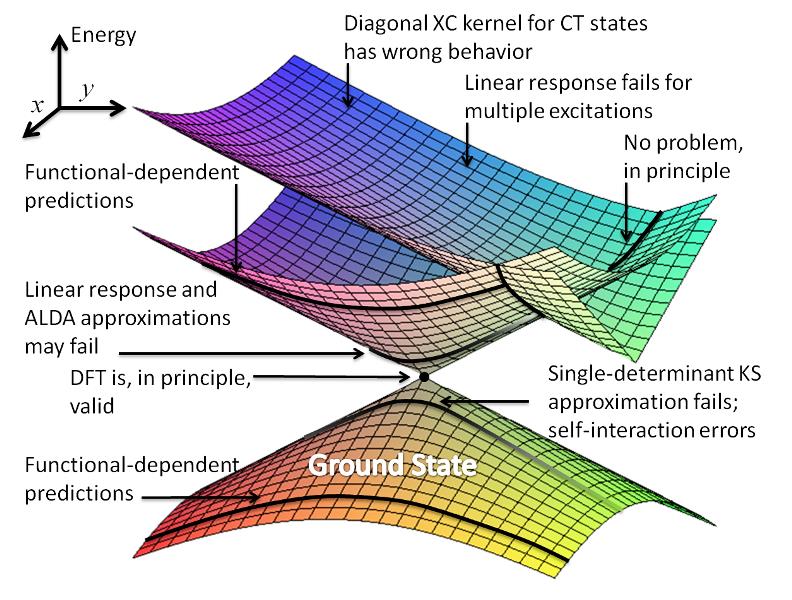
The paper is still not available at the publisher, but you can already get a preprint at the link below.
- M. Barbatti, R. Crespo-Otero, Surface Hopping Dynamics with DFT Excited States, in Density-functional methods for excited states, edited by N. Ferré, M. Filatov, M. Huix-Rotllant, Top. Curr. Chem. in press (Springer, 2015). doi:10.1007/128_2014_605

1 Comment
Surface hopping with TD-DFTB | Light and Molecules · December 2, 2017 at 7:41 AM
[…] states within DFTB can be computed following the same formalisms as in DFT, such as the linear-response (LR) time-dependent (TD) approach, that is TD-DFTB; or even the […]
Comments are closed.