Nonadiabatic dynamics can predict the overall course of a photochemical reaction. However, quantitative accuracy remains challenging.
In brief:
-
Fifteen teams predicted the photochemistry of cyclobutanone and its ultrafast electron-diffraction signal before the experimental results were available.
-
The electronic-structure method strongly influenced the predicted reaction times, often more visibly than the choice of nonadiabatic dynamics algorithm.
-
The exercise establishes practical benchmarks for predictive simulations, while exposing unresolved problems in accuracy, observables, and reproducibility.
Nonadiabatic molecular dynamics is commonly used to explain experimental results once the data are known. That is useful, but it is not quite a prediction. A more demanding test is to calculate what an experiment will observe before it is performed.
This was the idea behind the cyclobutanone prediction challenge. Participating teams were asked to simulate the effects of gas-phase cyclobutanone absorbing ultraviolet light at 200 nm. They also had to predict the time-resolved signal to be measured by MeV-UED. This technique follows structural changes in a molecule on femtosecond timescales.
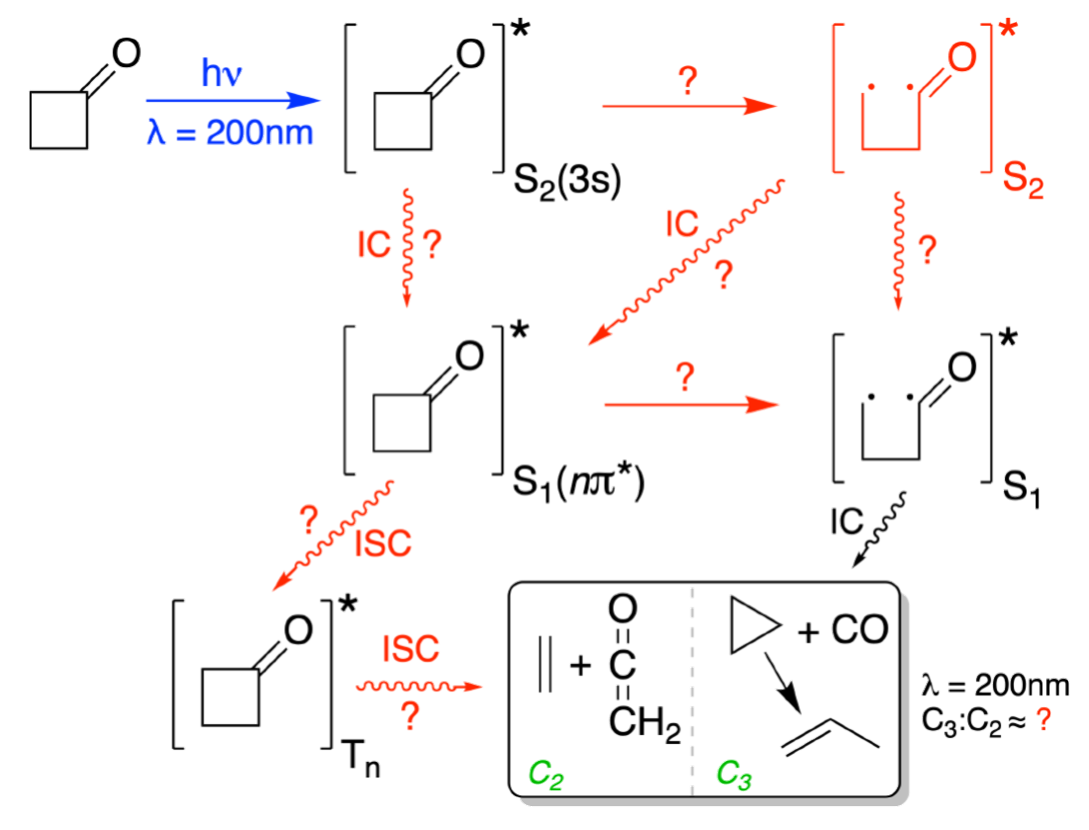
Fifteen predictions were submitted by more than 70 researchers using a broad range of electronic-structure theories, nonadiabatic dynamics methods, initial-condition protocols, and strategies for calculating the diffraction signal. The experimental results were obtained independently at facilities in Stanford and Shanghai.
In this community effort, led by Basile Curchod, we brought the predictions together and examined what could be learned from their agreements and disagreements. The analysis also incorporates discussions held during a dedicated CECAM workshop in Lausanne.
A reasonably consistent chemical picture emerged. The laser initially excites cyclobutanone to an electronic state with diffuse 3s Rydberg character. The molecule remains near this initial structure for a few hundred femtoseconds. It must then cross a small energy barrier, after which the excited state changes from Rydberg to valence character and the four-membered ring opens. Subsequent transitions to lower electronic states lead to products including carbon monoxide and cyclopropane or propene, as well as ethene and ketene. The calculations consistently indicated that intersystem crossing plays only a minor role under these excitation conditions.
That is the positive result. Nonadiabatic dynamics were qualitatively predictive. Many calculations anticipated the main photoproducts and reproduced the principal structural features observed in the diffraction experiment.
The quantitative picture was less reassuring. Predicted timescales and product yields varied substantially. Much of this variation could be traced to the electronic-structure method. Methods containing only static electron correlation generally produced ring opening too quickly. Methods dominated by dynamic correlation could trap the molecule near the initially excited structure for too long. Approaches combining static and dynamic correlation, particularly XMS-CASPT2, gave the closest overall agreement with the experimental timescale.
By contrast, the available results did not reveal a clear dependence on the family of nonadiabatic dynamics methods. This does not prove that all dynamics methods are equivalent. Too many other ingredients changed simultaneously for such a conclusion to be justified.
The challenge also exposed a weakness in the calculation of experimental observables. Most teams used an independent-atom approximation that omitted the inelastic-scattering feature associated with the Rydberg state. Predicting molecular dynamics is only part of the task; transforming those dynamics into the laboratory-measured signal can be equally demanding.
This work is therefore best viewed as a calibration exercise. It shows that predictive photochemistry is possible, but confidence requires electronic-structure benchmarks that extend well beyond the initial excitation geometry, explicit reporting of computational choices, better treatment of observables, and a serious assessment of uncertainty. The paper can be cited for its consolidated comparison of the fifteen predictions, its reconstruction of cyclobutanone photochemistry, and its practical recommendations for future nonadiabatic dynamics studies.
MB
Reference
[1] J. Janoš, N. H. List, A. J. Orr-Ewing, J. Suchan, M. Barbatti, O. Bennett, M. Brady, J. Carmona-García, R. Crespo-Otero, J. Eng, O. J. Fajen, M. Garavelli, S. Gómez, A. E. Green, F. J. Hernández, D. Hollas, L. Hutton, L. M. Ibele, A. Kirrander, Z. Lan, Y. Lassmann, J. E. Lawrence, B. G. Levine, D. V. Makhov, J. R. Mannouch, X. Miao, R. Mitrić, S. M. Parker, T. J. Penfold, J. Peng, J. O. Richardson, D. Shalashilin, P. Slavíček, K. E. Spinlove, P. Vindel-Zandbergen, F. Agostini, S. Bonella, T. J. Martínez, G. A. Worth, B. F. E. Curchod, Perspective on a challenge: Predicting the photochemistry of cyclobutanone, J. Chem. Phys. 165, 020902 (2026). 10.1063/5.0338792