A compact ML model trained on high-level multireference data enables nonadiabatic dynamics for hexatriene.
In brief:
-
Active learning turned a huge exploratory dataset into a small high-level one: about 850,000 AIQM1-sampled geometries were reduced to about 3,000 representative structures recomputed at MR-CISD level.
-
A machine-learning potential made MR-CISD-quality dynamics practical: the trained HIP-NN model allowed 200 nonadiabatic trajectories up to 800 fs, a scale that would be prohibitive with direct on-the-fly MR-CISD calculations.
-
The dynamics reveals a two-step relaxation mechanism: hexatriene relaxes mainly through S2→S1→S0, with localized S2/S1 hopping geometries, a broader S1/S0 seam, and early twisting around the central C=C bond.
All-trans-hexatriene is a small molecule, but it is not an easy one.
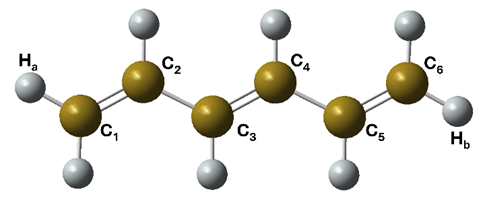
All-trans-hexatriene.
It belongs to the family of conjugated polyenes, molecules whose alternating single and double bonds make them sensitive to light. Such systems are useful benchmarks because they contain the same basic ingredients that appear in larger photoactive molecules: bright excited states, dark excited states, internal conversion, and possible photoisomerization. The difficulty is that these ingredients do not play nicely with cheap electronic-structure methods.
The central problem is the coexistence of ionic and covalent excited states. In simple terms, some excited states are well described by moving one electron from one orbital to another. Others need a more collective description, involving strong double-excitation character. A method that treats one class well may fail badly for the other. Multireference configuration interaction with singles and doubles, MR-CISD, is appropriate for this type of problem, but it is far too expensive for routine on-the-fly nonadiabatic dynamics.
In this work, led by Luan G. F. dos Santos, we asked whether machine learning could remove this bottleneck without throwing away the electronic-structure quality. The answer is cautiously positive.
The strategy was not to train the ML potential in two steps. First, we ran exploratory nonadiabatic dynamics with AIQM1, a much cheaper hybrid method that combines semiempirical quantum mechanics, a neural-network correction, and dispersion terms. These trajectories were not meant to provide the final answer. They were used as a map of the regions of molecular motion that hexatriene is likely to visit after light absorption. From this map, containing about 850,000 configurations. Then, we applied an active-learning procedure on them, which reduced the dataset to mere about 3,000 geometries. Only these selected geometries were then recomputed at the expensive MR-CISD level.
A HIP-NN machine-learning interatomic potential was then trained on this compact but carefully chosen dataset. This is the key point. The machine-learning model compressed expensive multireference information into a form fast enough for dynamics.
With this model, we propagated 200 trajectories up to 800 fs. The resulting dynamics gives a physically consistent picture: excitation starts in the bright S2 state, followed by sequential relaxation to S1 and then to the ground state. The computed S1 lifetime agrees well with gas-phase experimental measurements, while the simulated S2 decay is slower than experiment. That caveat matters. It points to remaining limitations in the electronic-structure treatment and in the dynamical model, rather than being hidden behind a good-looking population curve.
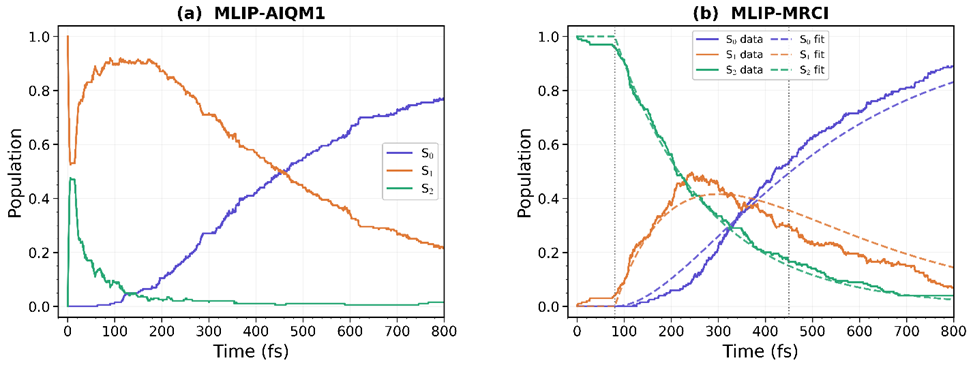
Time evolution of the adiabatic populations of hexatriene obtained from 200 surface hopping trajectories using (a) ML trained on AIQM1 and (b) ML trained on MRCI.
The structural analysis is also revealing. The S2→S1 hops occur in a relatively localized torsional region, mainly involving twisting around the central C=C bond. By contrast, the S1→S0 hops are much more diffuse. They spread over a broad portion of the intersection seam, rather than clustering around one neat geometry. The simulations also indicate that cis-trans isomerization starts mainly through motion around the central C3=C4 bond, although longer ground-state dynamics would be needed to determine final product ratios.
This paper should be cited for showing that active-learning machine-learning potentials can make multireference-quality nonadiabatic dynamics feasible for a challenging polyene benchmark, while still retaining mechanistic information about population flow, conical-intersection access, and early photoisomerization.
MB
Reference
[1] L. G. Fonseca Dos Santos, J. C. Chagas, M. Martyka, P. O. Dral, M. Barbatti, F. B. C. Machado, R. Messerly, H. Lischka, A Data-Efficient Machine-Learning Approach for Modeling the Photodynamics of All-trans Hexatriene based on Multireference Configuration Interaction Calculations, Faraday Discuss. (2026). 10.1039/D6FD00061D