Small chemical tweaks make a big difference: substitution patterns steer cinnamates toward faster nonradiative decay.
In brief:
-
We designed three methyl cinnamate derivatives to systematically accelerate excited-state deactivation.
-
Ultrafast spectroscopy (solution and gas phase) shows a dramatic acceleration upon adding a para-methoxy group, followed by steric hindrance at the C=C bond.
-
Multireference calculations map how these substitutions reshape the potential-energy landscape and conical intersections that “dump” energy back to the ground state.
E-Z photoisomerization is a key relaxation process in biological chromophores, but can we rationally redesign a classic photoisomerizable scaffold so that it converts absorbed light into heat as fast and as reliably as possible?
Nature already does this trick extremely well. In vision, the chromophore cis-11-retinal undergoes an ultrafast E–Z photoisomerization, and a key player in that speed is a conical intersection—a kind of “emergency exit” between electronic states that allows the molecule to return to the ground state without emitting light (i.e., by internal conversion, turning electronic energy into nuclear motion/heat).
Our target family was the cinnamates, widely found in plants and strongly associated with photoprotection and UV-filter chemistry. The design logic was two-pronged. First: use electron-donating substitution on the aromatic ring to tune electron density around the isomerizable double bond, with the hope of making the conical-intersection route more accessible. Second: use steric effects to encourage twisting about the C=C bond, because torsion is typically the geometric motion that carries the nuclear wavepacket toward the intersection.
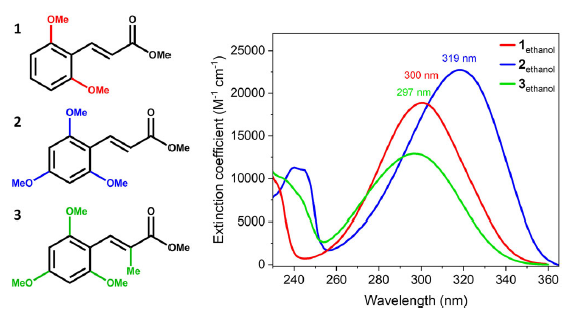
Methyl cinnamate derivatives. (Left) Molecules investigated in this work: (E)-di-ortho-methoxy-MC (1), (E)-di-ortho, para-methoxy-MC (2), and methyl-(E)-di-ortho, para-methoxy-MC (3). (Right) UV-visible absorption spectra for molecules 1-3 in ethanol, with measured band maxima labelled.
Our lab colleagues synthesized and studied three derivatives: (1) di-ortho-methoxy methyl cinnamate, (2) di-ortho plus para-methoxy, and (3) the same as (2) but with an added methyl substituent on the central double bond to pre-twist the chromophore. Then they attacked the dynamics from both sides: femtosecond transient absorption in solution and time-resolved ion-yield/photoelectron spectroscopy in the gas phase. The punchline is satisfying: each step in the substitution strategy accelerates the relaxation. The para-methoxy substitution collapses the excited-state lifetime by orders of magnitude compared with molecule (1), and adding steric hindrance at the double bond makes relaxation even faster—into a regime that approaches the celebrated speed of retinal isomerization.
Experiment alone tells you that the clock speeds up. To understand why, Josene Toldo and I mapped the excited-state landscapes using multireference electronic-structure methods (XMS-CASPT2 and MRSF-TDDFT), focusing on the topology and accessibility of key S₁/S₀ conical intersections. The story that emerges is nicely mechanistic: molecule (2) still supports a relatively stabilizing excited-state region (effectively a shallow “rest stop”) that introduces a small barrier on the way to the intersection, whereas molecule (3) becomes pre-twisted and lacks that stabilizing minimum—so the nuclear wavepacket can run downhill more directly toward the intersection, i.e., a more ballistic, barrierless return to the ground state.
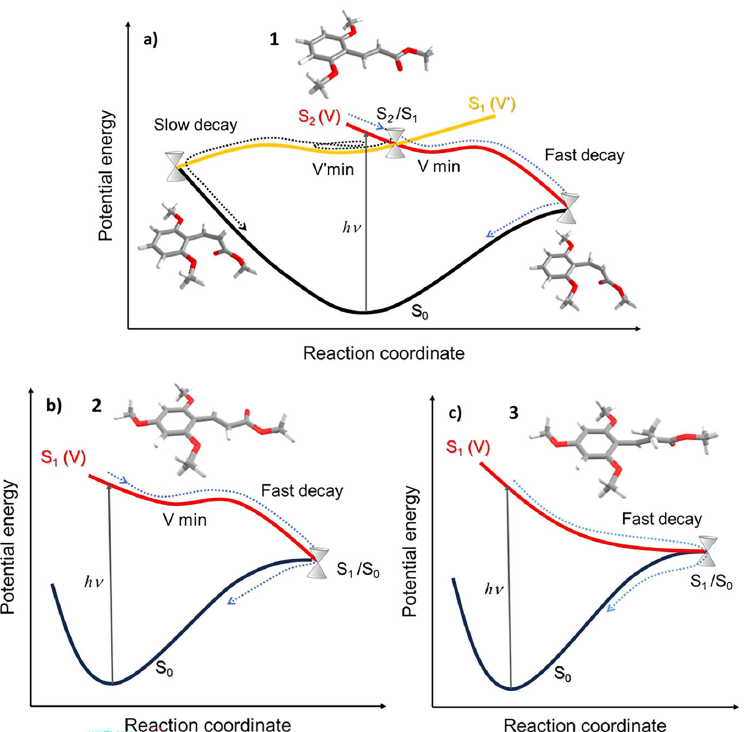
Schematic representation of the excited-state decay pathways proposed for molecules 1 (a), 2 (b) and 3 (c).
Beyond cinnamates, the broader point is design-oriented: by linking specific substitution patterns to specific potential-energy-surface features, you get a practical recipe for engineering efficient photon converters for heat generation, photo-switching, or light-filtering applications.
MB
Reference
[1] 1 M. Hymas, J. Dalton, I. Romanov, H. Sanders, M. Barbatti, J. M. Toldo, W. J. Buma, V. G. Stavros, Optimizing photon conversion routes in cinnamate derivatives, Commun. Chem. 9, 163 (2026).
DOI: 10.1038/s42004-026-01963-2