Methods based on nuclear ensembles allow to simulate steady and time-resolved photoelectron spectra with absolute intensities.
Providing results directly comparable to experimental data is a major goal in computational theoretical chemistry. This goal represents a special challenge with the natural trend of dealing with always larger and more complex molecular systems. Whenever nonlocal quantum effects can be neglected, resorting to semiclassical simulations turns out to be a good option, as it allows closely emulating experimental techniques at relatively modest computational costs compared to full quantum simulations.
In the last years, we have worked out and implemented diverse of such semiclassical approaches for spectrum simulations within the Newton-X platform. A central point in these developments has been to include inhomogeneous broadening via nuclear ensembles.
In the present work [1], Wilmer Arbelo-Gonzalez, Rachel Crespo-Otero, and I apply this approach once more to develop methods for simulation of steady and time-resolved photoelectron (PE) spectra. Our implementation allows:
- computation of intensities with absolute units
- simulations at TDDFT level using a Newton-X / ezDyson / Gaussian 09 interface
- possibility of integration with other electronic structure methods
- use of arbitrary ensembles (dynamics, Wigner distributions, etc.)
- use of two different models for intensity calculation
- use of two different models for vibrational overlap modulation
For both, steady and time-resolved PE spectra, we have developed and tested two approximations, one based on full computation of photoelectron cross sections (we call it cross section approach) and another based on cross sections approximated by Dyson orbital norms (DO norm approach).
Moreover, the vibrational modulation of the electron kinetic energy distribution was also modeled with two different approaches. In the first one, vibrational overlaps between N and N-1 electron systems were supposed to be significant only for the electrons ejected with the maximum allowed kinetic energy (PVB model). In the second approach, vibrational overlaps were supposed to be constant over the whole electron kinetic energy domain (CVB model).
We applied these methods to simulate PE spectra of imidazole (steady and time-resolved) and of adenine (steady). The comparison to experimental data shows that steady spectra can be nicely predicted with the PVB model, with good description of intensities and band shapes.
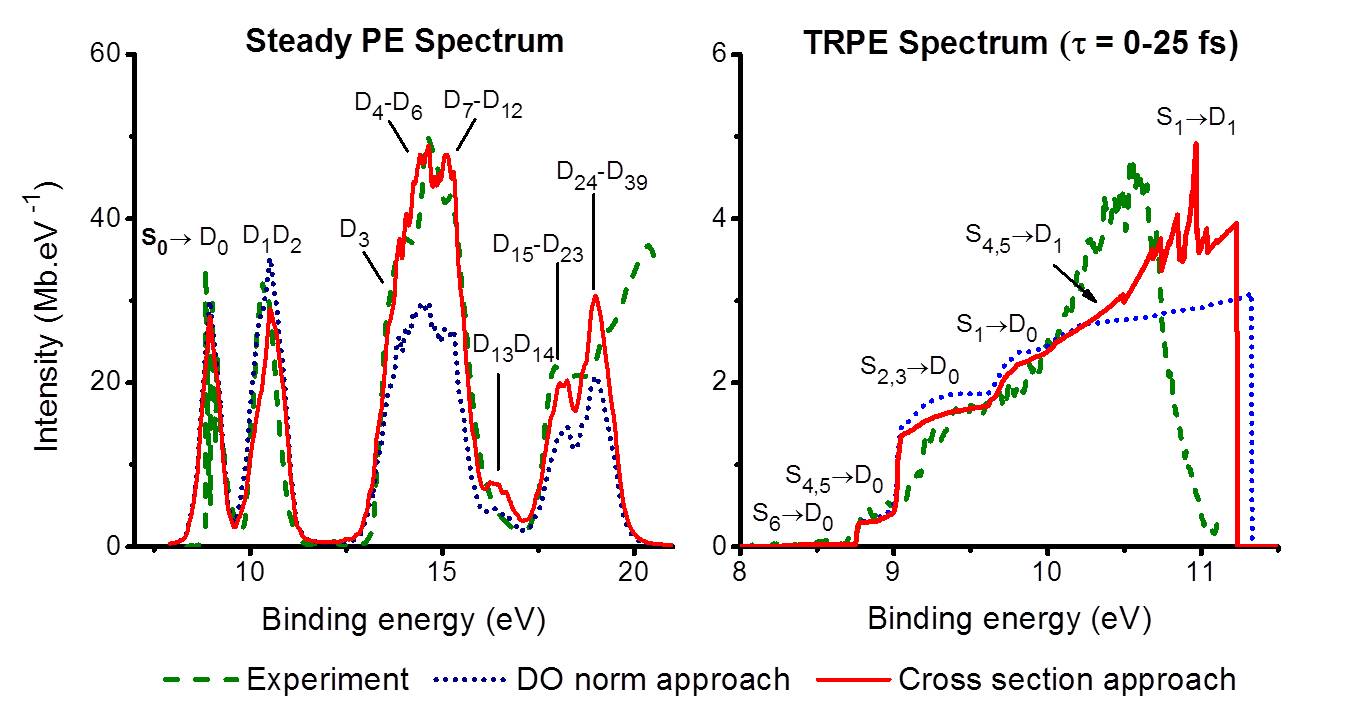
Steady (left) and time-resolved (right) PE spectra of imidazole. In the TRPE, only the initial time window is shown. Sn are the states of the neutral molecule, while Dn are the states of the cation.
For time-resolved spectra, however, the PVB model fails and the CVB model renders much better results. The CVB simulations have been able to reproduce a series of substructures in the experimental spectrum.
For all tested cases, cross sections approximated by Dyson orbital norms delivered results of similar quality as those based on full computation of cross sections, with an enormous economy of computational effort.
These results make us confident that photoelectron spectrum simulations based on the nuclear ensemble approach can be an effective tool to aid deconvolution and assignment of experimental data for large molecules.
The methods will be available in the next release of Newton-X.
Reference
[1] W. Arbelo-González, R. Crespo-Otero, and M. Barbatti, Steady and time-resolved photoelectron spectra based on nuclear ensembles , J. Chem. Theory Comput. doi: 10.1021/acs.jctc.6b00704 (2016) (50 free copies)