A new molecular set composed of small haloorganic compounds shows that popular computational methods may have errors of about 0.8 eV.
Everyone working on computational chemistry of excited states is used to errors at about 0.2 eV in energy estimates. This is the typical accuracy we get when comparing excited-state energies to reference experimental or high-level computational data.
This 0.2-eV accuracy has been the best we can meet with any of the most popular computational methods, ranging from TDDFT to CASPT2.
If this large error is already worrisome, things can go even worse.
The most popular molecular sets available in the literature overemphasize conjugated organic compounds, whose spectra are dominated by excitation of π electrons. But how do computational methods for excited-state simulations perform for non-conjugated haloorganic systems?
This question hit us recently when we started looking into the photochemistry of HCFC and brominated compounds. In spite of their importance for green-house effect and ozone depletion, we quickly realized that these substances have been completely neglected by computational chemists. We could not find any report on the accuracy of computational methods for this class of systems.
Aiming at closing this knowledge gap, we are introducing the Halons-9 molecular set [1].
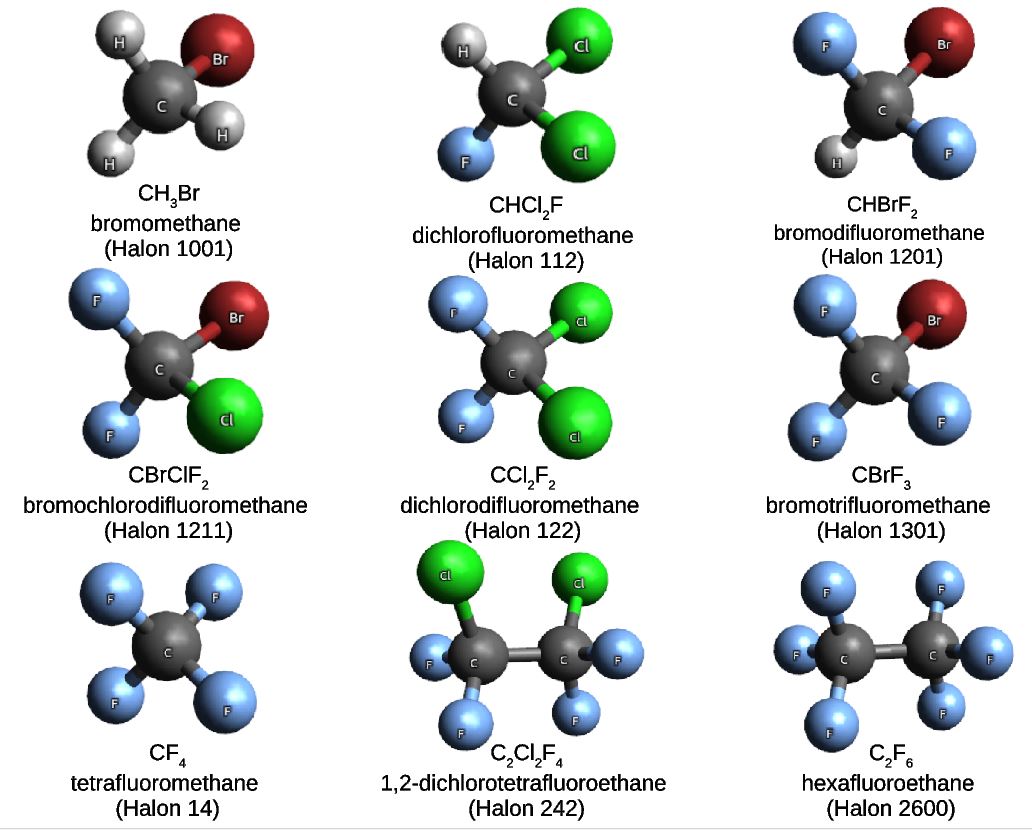
In a project led by Ljiljana Stojanovic, and in collaboration with our colleagues from the King Abdulaziz University in Jeddah, we built a molecular set composed of nine haloalkane compounds also known as halons. Halons are very well-known substances, often used in industrial applications. In particular, the nine halons that we have chosen for the Halons-9 molecular set are gaseous under normal conditions of temperature and pressure.
Given the general formula CαH(2α-β-γ-δ+2)FβClγBrδ, the Halons-9 molecular set includes the compounds listed in the Table and illustrated in the Figure below.
| Compound | α | β | γ | δ | Name |
| CH3Br | 1 | 0 | 0 | 1 | Halon-1001 |
| CHCl2F | 1 | 1 | 2 | 0 | Halon-112 |
| CHBrF2 | 1 | 2 | 0 | 1 | Halon-1201 |
| CBrClF2 | 1 | 2 | 1 | 1 | Halon-1121 |
| CCl2F2 | 1 | 2 | 2 | 0 | Halon-122 |
| CBrF3 | 1 | 3 | 0 | 1 | Halon-1301 |
| CF4 | 1 | 4 | 0 | 0 | Halon-14 |
| C2CL2F4 | 2 | 4 | 2 | 0 | Halon-242 |
| C2F6 | 2 | 6 | 0 | 0 | Halon-2600 |
As a first test with the Halons-9 molecular set, we computed excited states up to the first ionization threshold for these nine compounds with TDDFT (CAM-B3LYP), DFT/MRCI, and EOM-CCSD. Then, the states were assigned and classified as valence, Rydberg, and mixed valence-Rydberg.
Taking EOM-CCSD as the reference data set, the mean absolute error (MAE) considering all states is 0.8 eV for TDDFT and 0.5 eV for DFT/MRCI (always with the def2-TZVPP basis set).
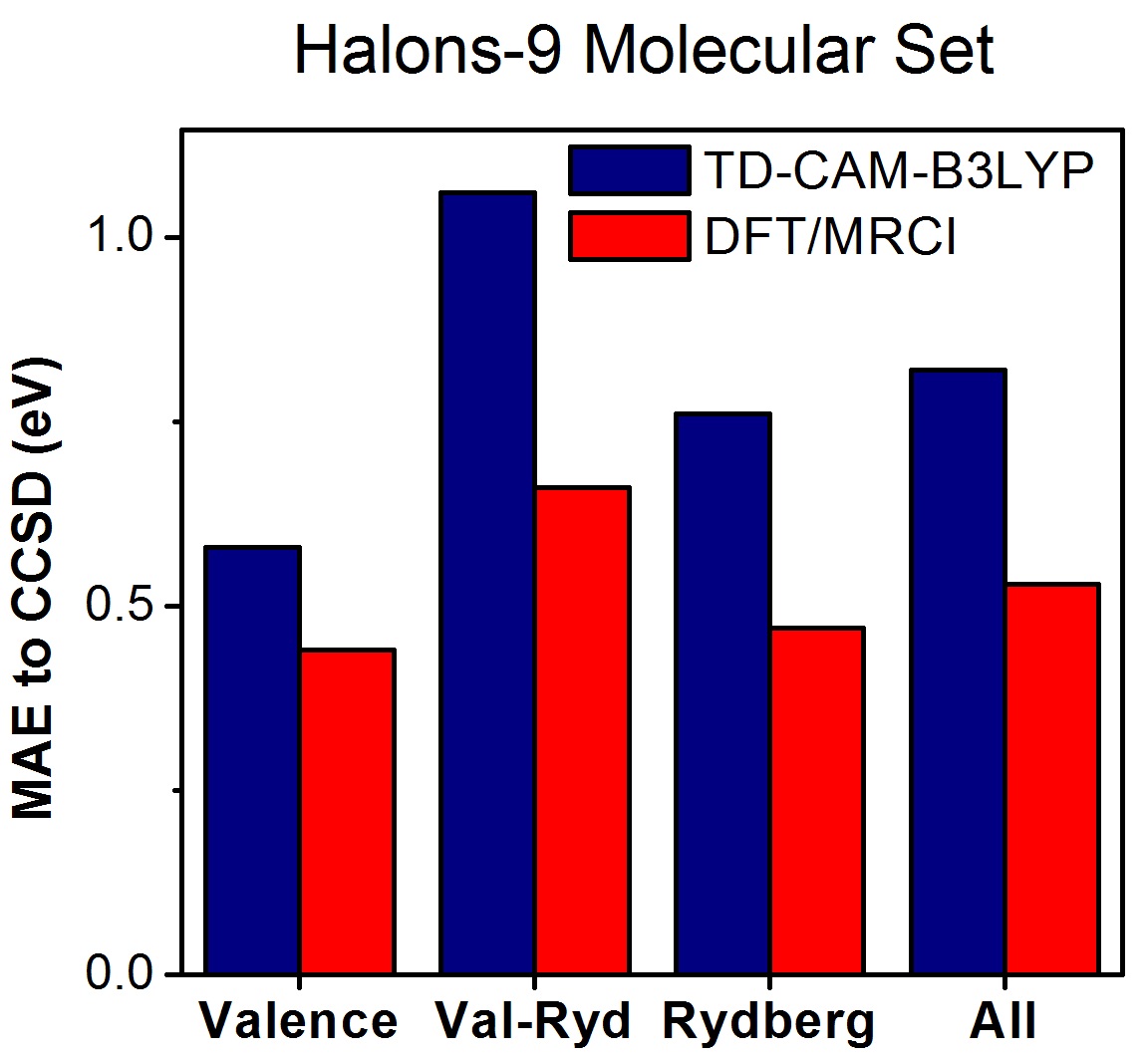
The strong deviation between DFT-based methods and CCSD is related to the wrong asymptotic behavior of the exchange-correlation (XC) potential (not of the functional, which is already corrected in CAM-B3LYP). This problem does not have a strong effect on low-energy excited states (as those of conjugated systems), but it strongly impacts high-energy states, which are dominant in the Halons-9 molecular set.
This first benchmark is focused on spin-free states. Nevertheless, a full account of how computational methods perform when simulating the Halons-9 molecular set should consider spin-orbit effects too; they are particularly important for compounds with bromine and chlorine atoms.
We hope that these critical results obtained for the Halons-9 molecular set will motivate further tests and methodological developments to deal with this class of compounds.
Reference
[1] L. Stojanovic, A. Alyoubi, S. Aziz, R. H. Hilal, and M. Barbatti, UV Excitations of Halons, J. Chem. Phys. 145, 184306 (2016). doi: 10.1063/1.4967170


