Better electron correlation can improve nonadiabatic dynamics—but it also introduces new numerical fragilities that we can’t ignore.
In brief:
-
Fully correlated methods are most useful when chemistry occurs (bond-breaking/formation, hot ground-state dynamics), not necessarily for routine excited-state relaxation.
-
They are not a magic upgrade button: active-space instabilities, discontinuities, and convergence issues can derail trajectories.
-
These are conclusions from a benchmark of 17 surface-hopping simulation batches on fulvene and pyrrole across seven electronic-structure families.
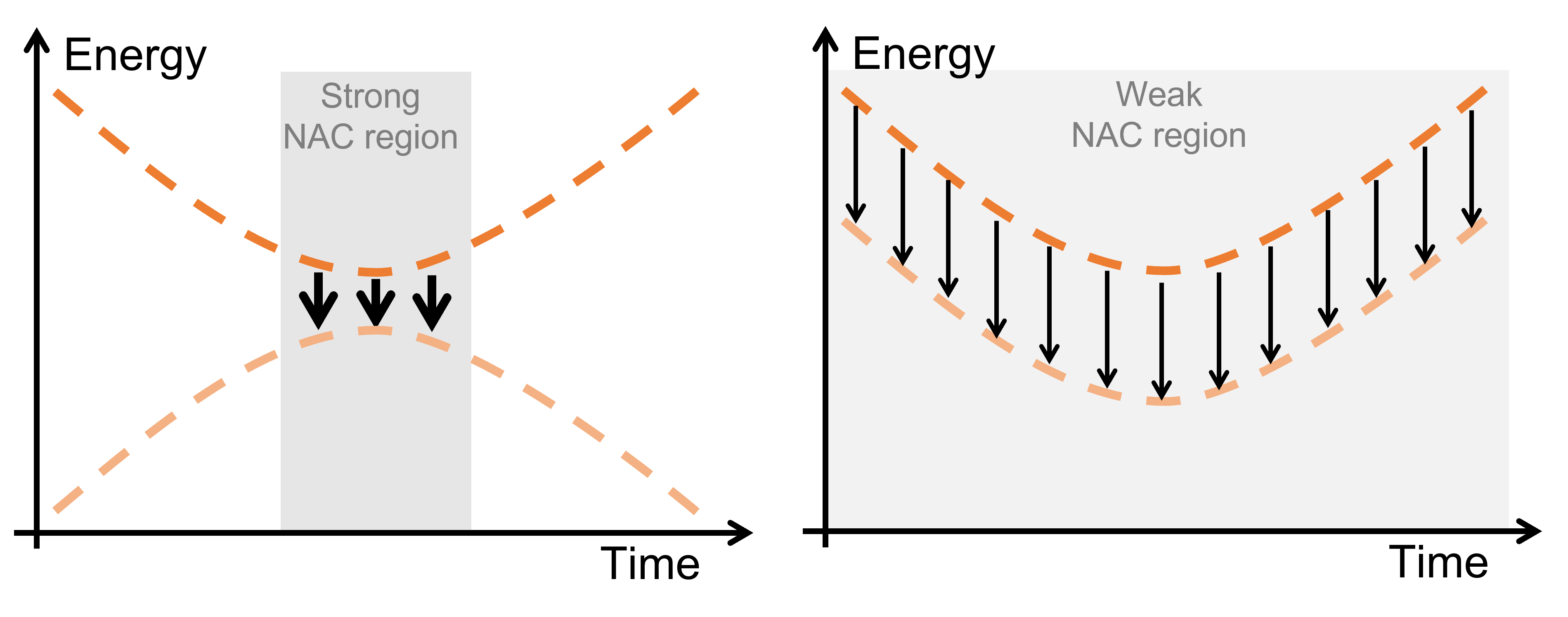
Surface hopping has become the practical workhorse for simulating ultrafast photochemistry. However, the cost is brutal: every trajectory needs electronic energies, gradients, and couplings at every step, so you end up doing an absurd number of quantum-chemistry calculations in a single study. That computational pressure explains why so much of the literature relies on partially correlated methods—things like CASSCF (mostly static correlation) or TDDFT and ADC(2) (mostly dynamic correlation).
But we’re clearly entering a new phase. “Fully correlated” approaches—here meaning methods that aim to balance static and dynamic correlation (not “exact correlation”)—are increasingly available for dynamics: XMS-CASPT2, L-PDFT, and MRSF-TDDFT are the main protagonists in our story.
The Perspective makes two moves. First, we try to clean up the conceptual foundations. A surprisingly persistent misconception is that dynamic electron correlation involves electrons “moving around in time.” It doesn’t. Correlation energy is defined within the time-independent Schrödinger equation as the difference between the exact nonrelativistic energy and the Hartree–Fock mean-field energy; time is not a variable in that definition.
Second—and this is where we went beyond the usual Perspective vibe—we ran 17 batches of decoherence-corrected fewest-switches surface hopping (DC-FSSH) simulations for two test molecules with very different profiles: fulvene (a proxy for photophysical relaxation and lifetime problems) and pyrrole (a classic case where photochemistry and bond rearrangement force us to confront static+dynamic correlation together).
 What comes out is not a victory lap for the most expensive method. Fully correlated approaches can improve the description of bond rearrangements and hot ground-state dynamics, but they also expose new failure modes: active-space orbital instabilities, potential-energy discontinuities, intruder-state issues (for CASPT2), and plain-old convergence crashes. Meanwhile, for comparatively gentle observables—like excited-state lifetimes and dynamics away from S₁/S₀ seam—ADC(2) and TDDFT can be entirely adequate, sometimes preferable in practice, because they tend to be cheaper and more numerically robust.
What comes out is not a victory lap for the most expensive method. Fully correlated approaches can improve the description of bond rearrangements and hot ground-state dynamics, but they also expose new failure modes: active-space orbital instabilities, potential-energy discontinuities, intruder-state issues (for CASPT2), and plain-old convergence crashes. Meanwhile, for comparatively gentle observables—like excited-state lifetimes and dynamics away from S₁/S₀ seam—ADC(2) and TDDFT can be entirely adequate, sometimes preferable in practice, because they tend to be cheaper and more numerically robust.
So the conclusion is annoyingly grown-up: method choice remains problem-dependent. The path forward is likely a mix of more robust multireference machinery (generalized active spaces), better integration of electronic-structure and dynamics, and complementary strategies (models, fits, ML potentials) rather than a single ultimate electronic-structure ladder rung.
MB
Reference
[1] E. G. F. de Miranda, R. Souza Mattos, S. Mukherjee, J. M. Toldo, C. H. Choi, M. T. do N. Varella, M. Barbatti, Surface Hopping with Fully Correlated Methods, J. Chem. Theory Comput. (2025). 10.1021/acs.jctc.5c01529