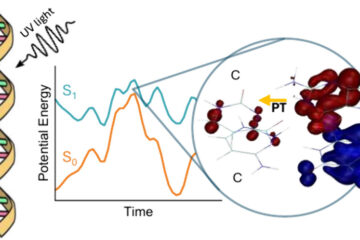
Benchmark of excited-state nonadiabatic dynamics simulations for adenine shows the limits of quantum chemical methods.
In a recent publication [1], we present the new implementation of surface-hopping dynamics in Newton-X using CC2 and ADC(2) from Turbomole and TDDFT from G09. We looked at the dynamics of 9H-adenine in the gas phase, as a benchmark case.
Confirming our previous investigations, TDDFT does not predict the ultrafast deactivation of this molecule. ADC(2), however, did quite a god job and it is so far the most reliable method that we tested in this benchmark. CC2, on its turn, was too unstable for excited-state dynamics.
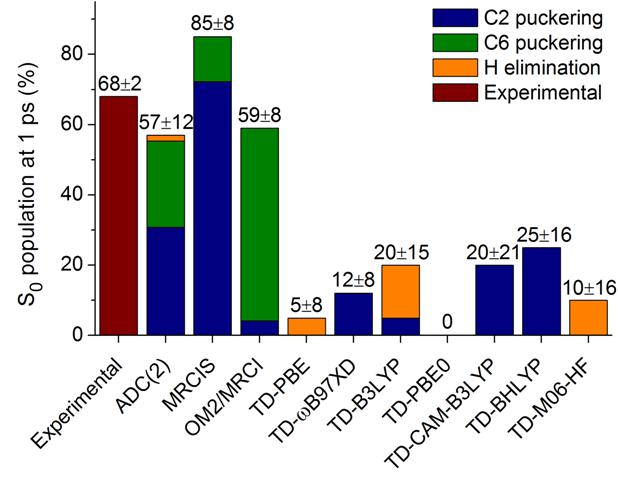
Excited-state dynamics of adenine with several different methods.
References
[1] F. Plasser, R. Crespo-Otero, M. Pederzoli, J. Pittner, H. Lischka, M. Barbatti, Surface hopping dynamics with correlated single-reference methods: 9H-adenine as a case study; JCTC, doi:10.1021/ct4011079 (2014).
doi:10.1021/ct4011079