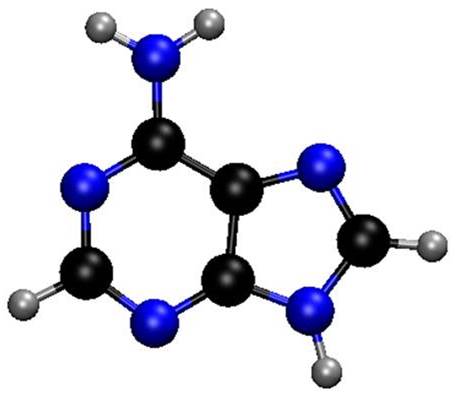
Using computational simulations of 2-aminopurine-water clusters, we show why a single water may turn the fluorescence of this molecule on.
Structurally, 2-Aminopurine (2Ap) is analogous to adenine (Ade). Photochemically they couldn’t be more different. After UV excitation, their excited-state lifetimes are completely divergent: In water, Ade is dark (as it is in the gas phase too); but 2AP is a bright fluorescent molecule, with a lifetime 100 thousand times longer (!) than that of Ade.
To be fair, the behavior of 2AP in water diverges even from its own behavior in the gas phase. In solution, 2Ap lifetime is still a stunning 600 times longer than that of isolated 2Ap.
| Excited-State Lifetime (ps) After 267 nm Excitation |
||
 |
 |
|
| 9H-Adenine | 2-Aminopurine | |
| Gas Phase | 1 | 30 |
| Water | 0.18 | 18.000 |
These photophysical idiosyncrasies of the two molecules have attracted the attention of researchers for a long time. Why does displacing the NH2 group from a carbon atom to another have such an impact? Why does water trigger the fluorescence in 2AP’s, but not in Ade?
Last year, in a series of impressive experiments based on time-resolved spectroscopy of microsolvated 2Ap, Lobsiger and co-workers [1] showed that the lifetime of 2AP increases systematically with the number water molecules. Even more significant, they also found out that position matters: for the same number of water molecules surrounding 2AP, the lifetime is much longer if one water interacts with the NH2.
Stimulated by these experiments, Hans Lischka and I decided to look at this problem using quantum chemistry. We investigated the photophysics of 2Ap-water clusters with up to 4 water molecules [2].
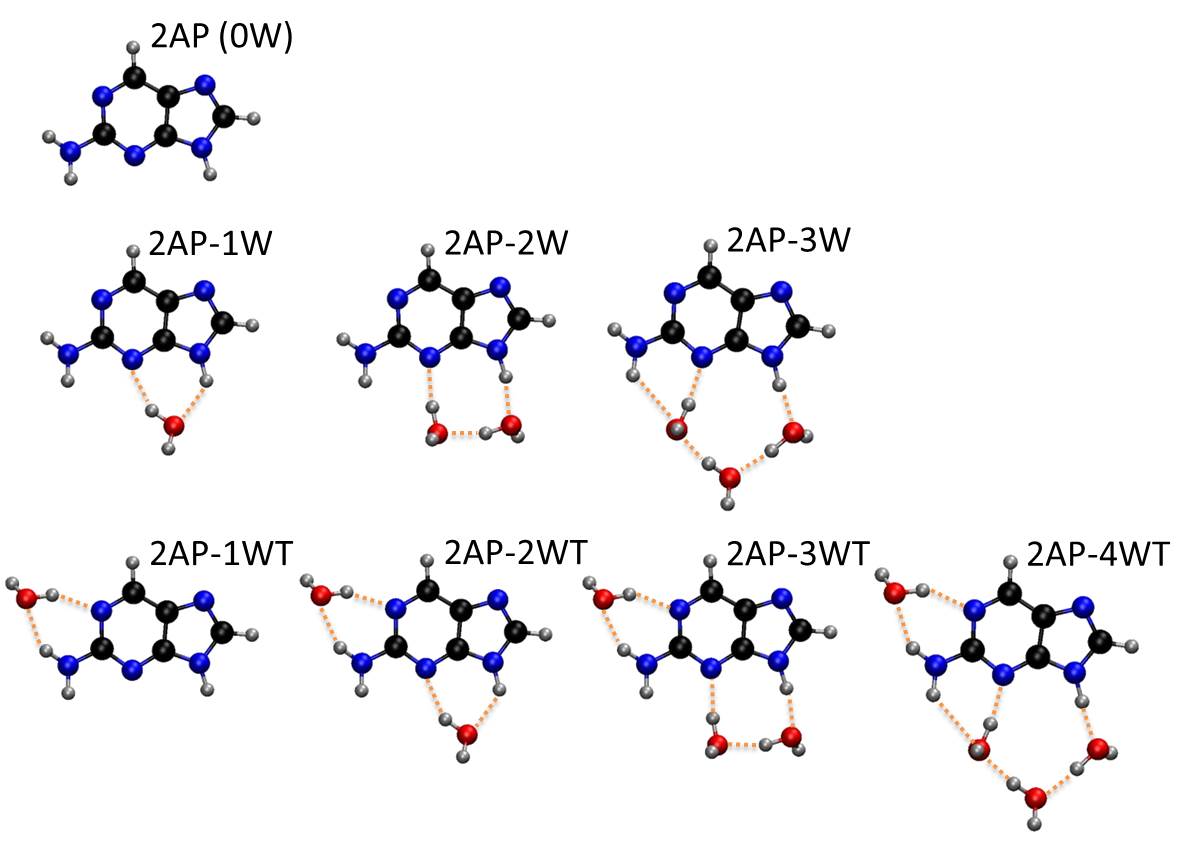
Investigated clusters at ADC(2) level.
Using the ADC(2) method, we showed that the dependence of the excited-state lifetime of 2AP on the number and position of water molecules is controlled by a barrier between the S1 minimum and a conical intersection where internal conversion can take place. Other possible competing pathways (proton transfer, intersystem crossing, and internal conversion at other intersections) were also investigated but discarded.
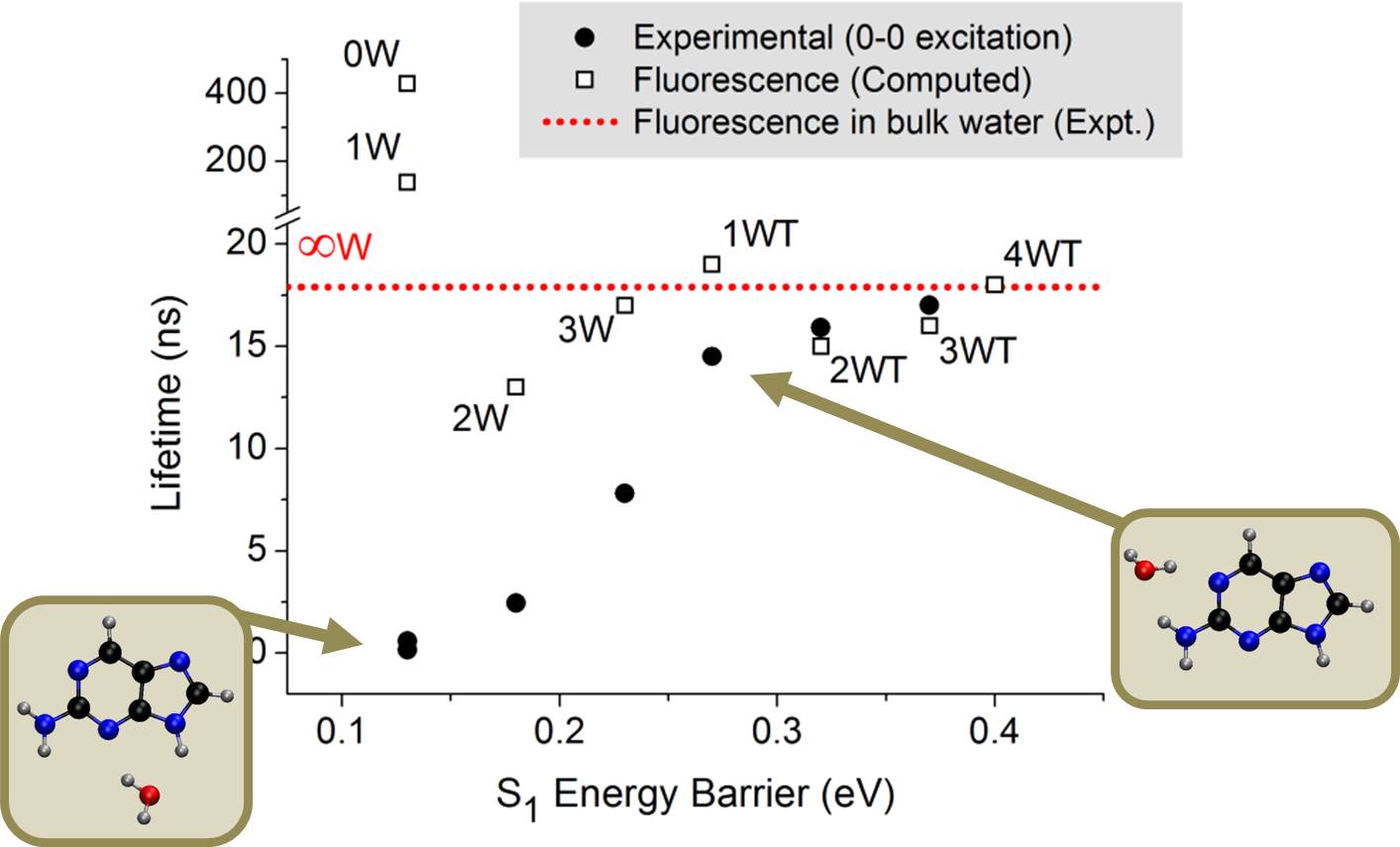
Experimental lifetimes after 0-0 excitation [1] as a function of the computed energy barrier for internal conversion [2]. The lifetime for the clusters with one water molecule can be either 0.6 ns or 15 ns, depending on where water attaches.
In vapor or when there are no water molecules interacting with the NH2 group, the connection between the S1 minimum (nπ*) and the conical intersection (nπ*/S0) is adiabatic, with a very small barrier. Thus, internal conversion rate is enhanced, lifetime is reduced, and fluorescence quenched.
On the other hand, when even a single water molecule interacts with the NH2 group, the pathway from the S1 minimum (now ππ*) to the conical intersection (still nπ*/S0) requires a nonadiabatic change, thus raising the energy barrier for internal conversion, increasing the lifetime, and enhancing the fluorescence.
Although there is still a long way to go as to understand the photochemistry of Ade and 2Ap, this interplay between advanced spectroscopy and high-level theory is clearly the way to fill the gaps.
References
[1] S. Lobsiger, S. Blaser, R. K. Sinha, H.-M. Frey, and S. Leutwyler, Switching on the fluorescence of 2-aminopurine by site-selective microhydration, Nat Chem 6, 989 (2014).
[2] M. Barbatti and H. Lischka, Why Water Makes 2-Aminopurine Fluorescent, Phys. Chem. Chem. Phys. doi:10.1039/C5CP01151E (2015).