A new approach based on dynamics simulations is proposed to boost the efficiency of organic photovoltaics.
Organic photovoltaics are inefficient. Their ability of converting light into electric current is only a fraction from that of silicon-based solar cells. But the many chemical, economical, and environmental advantages of working with organic materials for photo-energy conversion have motivated a legion of scientists to try to improve their performance.
To help with this task, Daniele Fazzi, Walter Thiel and I have just developed a new computational approach [1], where energies, relative ordering, and population of productive and unproductive electronic states of a molecular aggregate are monitored during the relaxation following the photoexcitation. (Productive states are those contributing to electric current generation, while unproductive states are those leading to charge recombination and dissipation of the light energy as heat. To know whether a state is productive or unproductive we use the state classification procedure that we have introduced before [2], with the electronic densities obtained during a TDDFT surface hopping dynamics [3].)
Applying this approach to a particular system—the photoexcited bi-thiophene dimer—we have shown that few femtoseconds after the light absorption, the dimer shows two electronic energy bands separated by a sizable energy gap. The high-energy band has a productive state lying on its bottom, while the low-energy band is composed of unproductive states only.
As the dimer relaxes, it populates the productive state on the bottom of the high-energy band. If the gap separating the bands remained constant, this productive state would survive for a long time, contributing to the current generation. However, induced by molecular vibrations, the energy gap fluctuates and, when it gets to a minimum, the population is transferred to the low-energy unproductive band. As result, the productive state survives for not more than 100 fs, rendering poor power conversion performance.
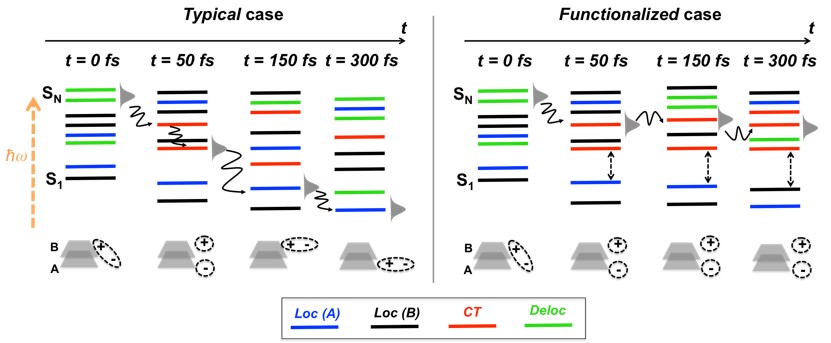
To increase the efficiency of the solar cell, the aim is to keep the “red” productive states populated as long as possible.
The vibrational modes responsible for shrinking the energy gap are the C-S stretching modes in the thiophene units. When these modes are frozen during the simulations, the low-energy band does not get populated and the lifetime of the productive state is substantially elongated.
Thus, this dynamic analysis of the electronic states teaches that, in the case of organic solar cells based on bi-thiophene dimers, we may be able to increase their power conversion efficiency if we find ways to restrict C-S vibrations.
In organic photodevices based on other molecules, the vibrational modes controlling the productive states’ population may be completely different. However, the approach that we have developed is absolutely general. It will always allow to find where the productive states are as a function of time. And with this information, we may devise ways to functionalize the molecule to boost the lifetime of productive states.
References
[1] D. Fazzi, M. Barbatti, W. Thiel, Unveiling the role of hot charge-transfer states in molecular aggregates via nonadiabatic dynamics, J. Am. Chem. Soc., doi: 10.1021/jacs.5b13210 (2016).
[2] K. Sen, R. Crespo-Otero, O. Weingart, W. Thiel, M. Barbatti, Interfacial states in donor-acceptor organic heterojunctions: computational insights into thiophene-oligomer/fullerene junctions; J. Chem. Theory Comput., 9 533 (2013). doi:10.1021/ct300844y
[3] M. Barbatti, R. Crespo-Otero, Surface Hopping Dynamics with DFT Excited States, Top. Curr. Chem. 368, 415 (2015). doi:10.1007/128_2014_605
1 Comment
Hot and cold charge transfer in organic donor/acceptor interfaces | Light and Molecules · September 18, 2017 at 2:38 PM
[…] If you want to know more about the dynamics at homogeneous interfaces, check: Organic photovoltaics, make light-not-heat […]
Comments are closed.