Attaching an electron donor group to the chromophore can speed up electron transfer.
In brief:
- Electron transfer (ET) rates from a photocatalyst to the substrate were computed for a metal-free organic reaction.
- The rates were determined for many different chemical substitutions in the photocatalyst.
- Electron-donating groups enhanced ET while electron-withdrawing groups blocked it.
Metal-free photocatalysts for organic synthesis are promising alternatives to conventional photocatalysts, as they may be inexpensive for scaling-up production, easily tunable for light adsorption, structurally flexible, and environmentally friendly. These features have made metal-free organic photocatalysts appealing in recent years, drawing increasing attention in a vibrant research field.
A crucial step to generate reactive species in the photocatalytic reaction is to boosting electron transfer (ET) from catalysts to substrates. In this project [1], led by Guozhen Zhang from Hefei, our goal was to apply computational chemistry to check how much the ET rates depend on the type of functional groups attached to a metal-free photocatalyst.
We did that for a well-established metal-free photocatalytic reaction, the pyrene-based (Py) hydrodefluorination of polyfluoroarenes (FA). The ET rates from Py to FA (C6F6) were systematically computed for a series of Py functionalized with different electron-donating (EDG) and electron-withdrawing groups (EWGs). Thus, our model photocatalyst was a Py(X4), where X = –H, –OH, –NH2; –CH=CH2, –C≡C–CH3, –C≡C–C(CH3)3; –NO2, –CN, and –CHO.
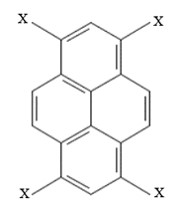
In practical terms, we computed the ET rate for the reaction
[Py(X4)]–-(FA)2 → Py(X4)-[FA]–[FA]
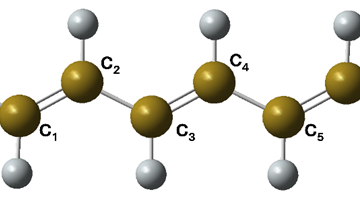
The molecular cluster structure of the simplest case, X = H, is shown in the figure below.
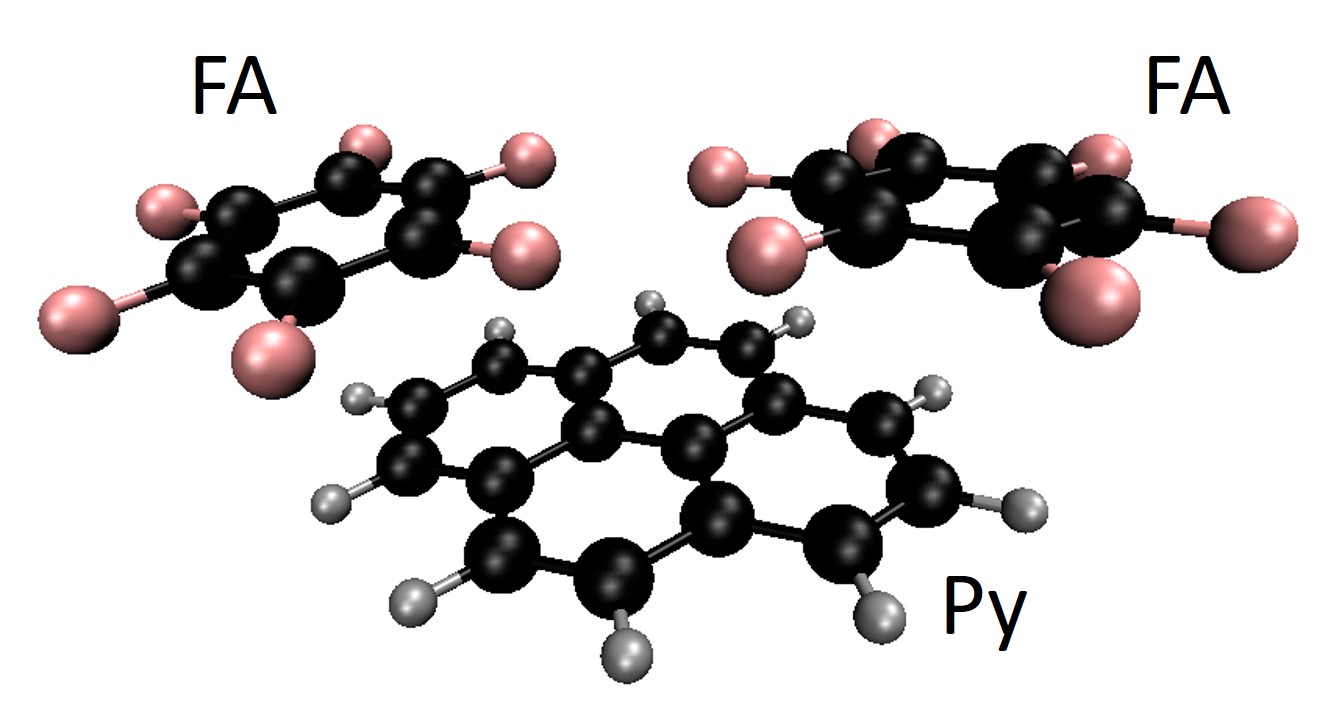
To compute the rates, we needed diabatic states with electrons localized either in Py or in FA. We got them through constrained DFT calculations. Then, we built 1-D potential energy profiles along the ET coordinate (corresponding to the bending of one of the F atoms toward Py).
For each X, the 1-D potential energy profiles rendered two low-frequency parabolic potentials, one for the electron localized in Py(X4) and another for the electron localized in FA. With these diabatic potentials, we could estimate the ET rate using semiclassical Marcus theory.
The absolute and relative values of the rates are given in the table below.
| Py(X4)-(FA)2 |
kET / s–1 | kREL |
| –H | 7.28×106 | 1 |
| –OH | 1.73×109 | 2.37×102 |
| –NH2 | 3.96×1010 | 5.44×103 |
| –CH=CH2 | 5.13 | 7.05×10-7 |
| –C≡C–CH3 | 8.44 | 1.16×10-6 |
| –C≡C–C(CH3)3 | 1.90×10-3 | 2.61×10-10 |
| –CN | 1.61×10-11 | 2.21×10-18 |
| –CHO | 2.09×10-10 | 2.87×10-17 |
| –NO2 | 1.24×10-18 | 1.71×10-25 |
The estimated ET rates show that they are appreciably enhanced by EDG substitutions (–OH, –NH2) and strongly weakened by EWG substitutions (–CN, –CHO, –NO2).
Noticeably, among the three quantities used to get the rate—state coupling, reorganization energy, and Gibbs free energy change between the minimum of each state—only the latter matters for modulating the substantial rate variation for different X. Thus, the Gibbs free energy change can be taken as a proxy to estimate the relative ET rate in other compounds.
Our findings of how ET rates are tunned for Py-FA complexes via chemical group modifications should help the design of metal-free photocatalysts for organic transformations, by providing a rationale of how reactive species can be generated. Moreover, our results establish the basic quantity that should be checked to estimate how efficient this process should be.
MB
Reference
[1] R. Liu, L. Yang, T. Yang, Y. Huang, M. Barbatti, J. Jiang, and G. Zhang, Modulating Electron Transfer in an Organic Reaction via Chemical Group Modification of the Photocatalyst, J. Phys. Chem. Letters, DOI: 10.1021/acs.jpclett.9b01970 (2019).