Content
- Nonadiabatic phenomena
- Ethylene and substituted ethylene
- Heterocycles
- Formamide
- Protonated Schiff Bases
- Azobenzene & azomethane
- Singlet O2 generation
Nonadiabatic phenomena
As a consequence of a photoabsorption (PA) in the UV or visible region of the spectrum, a molecule is activated to an excited electronic state. Immediately after the excitation, it starts to relax by following many different deactivation mechanisms.
The molecule can undergo an internal conversion (IC) to other states of same spin multiplicity or make an intersystem crossing (ISC) to a state of different multiplicity. From the excited state minima, it can return to the ground state either by the emission of a photon through fluorescent (Fl) and phosphorescent (Ph) processes, or it can deactivate without emitting radiation via internal conversion. The crossing between states where the internal conversion occurs is called a conical intersection.
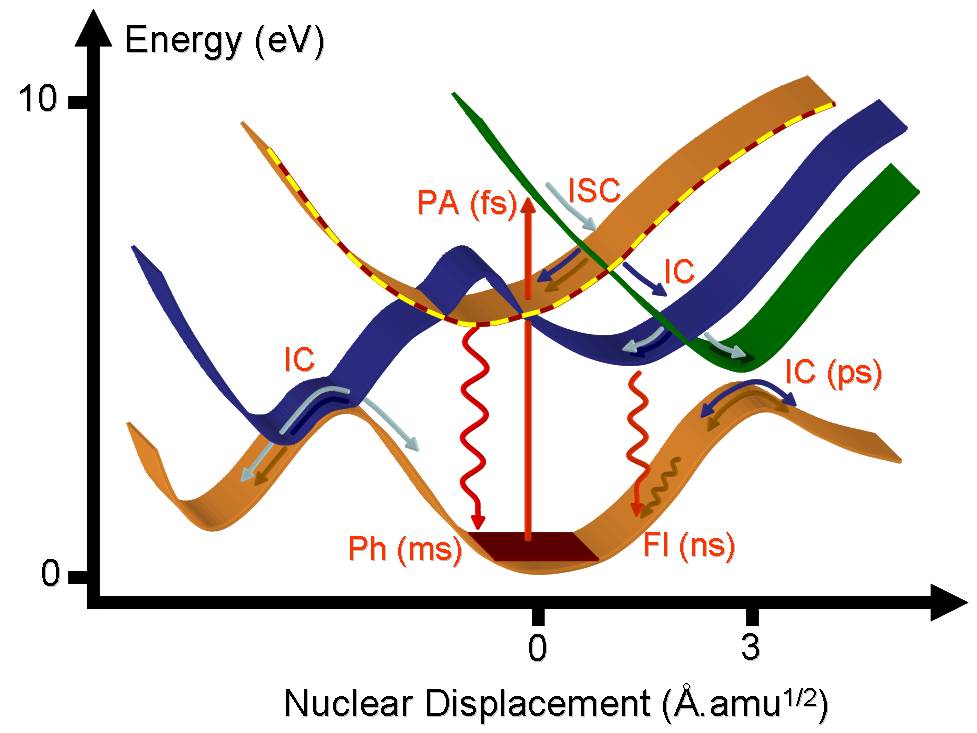
Schematic illustration of the deactivation mechanisms in a UV/vis excited molecules.
Each of these processes occurs at different time scales. The fastest, the internal conversion, may occur within few picoseconds (10-12 s) and usually involves conformers for which the Born-Oppenheimer approximation breaks down. The slowest, phosphorescence, may take microseconds (10-6 s). The preferential processes dominating each molecule depend upon the specific shapes of the excited state potential energy surfaces.
Based on our dynamics simulations results, we have reviewed in this paper how the internal conversion takes place in small organic molecules.
As a particularly relevant result, we have shown that internal conversion can be induced by an electron transfer from the solvent to the chromophore, a pathway that has never been reported before.
Ethylene and substituted ethylenes
The internal conversion in many organic molecules occurs by excitation of a ππ* state in a set of double bonds. For this reason, it is very important to understand how the internal conversion takes place in the simplest models for a double bond, ethylene and substituted ethylenes.
We have performed quantum chemical and excited state dynamics investigation of few of these small molecules, including ethylene (CH2=CH2) itself (Ref,Ref,Ref), fluoroethylene (CH2>=CHF) (Ref), silaethylene (CH2=SiH2) (Ref), and methaniminium cation (CH2=NH2+) (Ref,Ref).
These investigations have revealed that different from what is generally believed, the deactivation is usually not done through a single pathway, but through many deactivation pathways driving the molecule to different conical intersections.
Heterocycles
Any molecule has conical intersections connecting the excited and the ground state. Whether these intersections will be used for internal conversion or the molecule will deactivate by the emission of a photon depends only on how are the shapes of the surfaces of potential energy in the excited states.
Pyridone is an example of a fluorescent species which does not access its conical intersection. Following the experimental results of Leutwyler and co-workers, our simulations have shown that if a bias towards the out of plane vibrational modes is added right in the beginning of the dynamics, the conical intersections can be reached and the internal conversion rate increases (Ref).
Pyrrole (Ref,Ref,Ref,Ref) and imidazole (Ref) are non-fluorescent five-membered heterocycles. Using excited-state dynamics simulations, we have shown the statistical importance of each of the several deactivation pathways available for these molecules.
An important class of heterocycles undergoing internal conversion is the DNA/RNA nucleobases. Their ultrafast deactivation may represent a natural protection of the genetic code against UV excitation. The dynamics of nucleobases and base models are discussed in “Internal conversion of DNA/RNA nucleobases” page.
Formamide
Formamide is the simplest model for a peptide bond, which binds together the amino acids in a protein. For this reason, it is interesting to understand how it reacts to the UV excitation. Headed by our collaborators in Zagreb (M. Eckert-Maksic, I. Antol, and M. Vazdar), we have investigated the dynamics of formamide (Ref,Ref), protonated formamide (Ref), and of formamide in an Ar matrix (Ref). The multiple reaction pathways and their statistical importance have been mapped in each case.
In a recent collaboration with the experimental group of W. Sander (Ref), we have shown that the major product of the dissociation dynamics of methyl-formamide in Ar matrix is a weakly-bound complex between CH3NH and HCO radicals. This complex owes its existence to the cage effect of the matrix which allows for H-transfer reactions and recombination.
Protonated Schiff bases
It has been determined in the 1960’s that the initial molecular steps are the absorption of light by retinal, the chromophore of the rhodopsin, a protein present in the retina, which isomerizes as a consequence of the excitation.
Since then, the detail of the isomerization cycle of rhodopsin and other similar photoreceptors have become one of the most active fields of research in photochemistry and photobiology. The simplest models for retinal are the protonated Schiff bases known as PSBn. We have investigated the nonadiabatic dynamics of PSB3 (Ref) and PSB4 (Ref) by ab initio methods, under a variety of environmental restrictions. One of our main findings is the “Folding Table” isomerization mechanism, which takes place in PSB3 and PSB4 under a large degree of restriction. This mechanism is shown in the movie below.
Azobenzene & Azomethane
Azobenzene (Ph-N=N-Ph) and azomethane (CH3-N=N-CH3) are prototypes of molecular phototriggers.
We have investigated the nonadiabatic cis-to-trans isomerization of azobenzene after S1 (n–π*) excitation, employing the fewest-switches surface hopping method based on ab initio methods (Ref). Azobenzene photoisomerization occurs purely as a rotational motion of the central CNNC moiety. Two nonequivalent rotational pathways corresponding to clockwise or counterclockwise rotation are available. The course of the rotational motion is strongly dependent on the initial conditions. The internal conversion occurs via an S0/S1 crossing seam located near the midpoint of both of these rotational pathways. Based on statistical analysis, we showed that the occurrence of one or other pathway can be completely controlled by selecting adequate initial conditions.
The excited-state dynamics of azomethane is an ultrafast process leading to a clean formation of CH3 radicals. We have investigated the details of the initial steps in this process. First, we characterized the UV spectrum of this molecule (Ref), and then the first picosecond of dynamics up to the internal conversion in the gas phase (Ref) and in solution (Ref).
Singlet O2 generation
Thionucleobases
The investigation of singlet oxygen—including its generation, reactivity properties, and physiological cytotoxicity—has been a popular subject for several decades because of its broad importance for chemical synthesis, environmental sciences, phototherapy, and physiology.
In many applications, the extra energy needed to generate the singlet species from the triplet O2 ground state is harvested from the excited states of a photosensitizer (PS). During this photosensitization process, singlet oxygen is generated through the reaction
1[3PS + 3O2] → 1[1PS + 1O2]
But there is a long story before getting to this reaction, involving several steps:
- PS photoabsorption: PS(S0) → PS(Sn)
- PS relaxation to triplet manifold: PS(Sn) → PS(T1)
- PS triplet state intrinsic decay (without forming 1O2): PS(T1) → PS(S0)
- Then, finally, 1O2 formation: 3PS + 3O2 → 1PS + 1O2
Taking thionucleobases as a prototypical PS, we have been studying each of these steps.
We started by looking at the photoabsorption and the relaxation to the triplet manifold, to explain how the thio-substitution affects the photophysics.
Then, we looked at the intrinsic decay. We proposed some general rules to control the 1O2 yield by simply controlling the intrinsic triplet decay ad explained the rate differences between thionucleobases and trinucleotides.
Before we could address the 1O2 formation, however, we had to work on methods. The PS-O2 system is a floppy and weakly-coupled complex. There are not many methods around that would allow estimating the reaction rate for such a system.
We solved that problem with the Divide to Conquer (DtC) model, a kinetic model allowing to compute reaction rates to floppy and weakly-coupled complexes.
We also implemented PySOC, a program to quickly compute spin-orbit couplings.
With the DtC model, we showed that the reaction rate for singlet oxygen formation is deeply dependent on the PS-O2 incidence direction. We also showed how the PS-O2 should interact to maximize this rate.
Porphycene
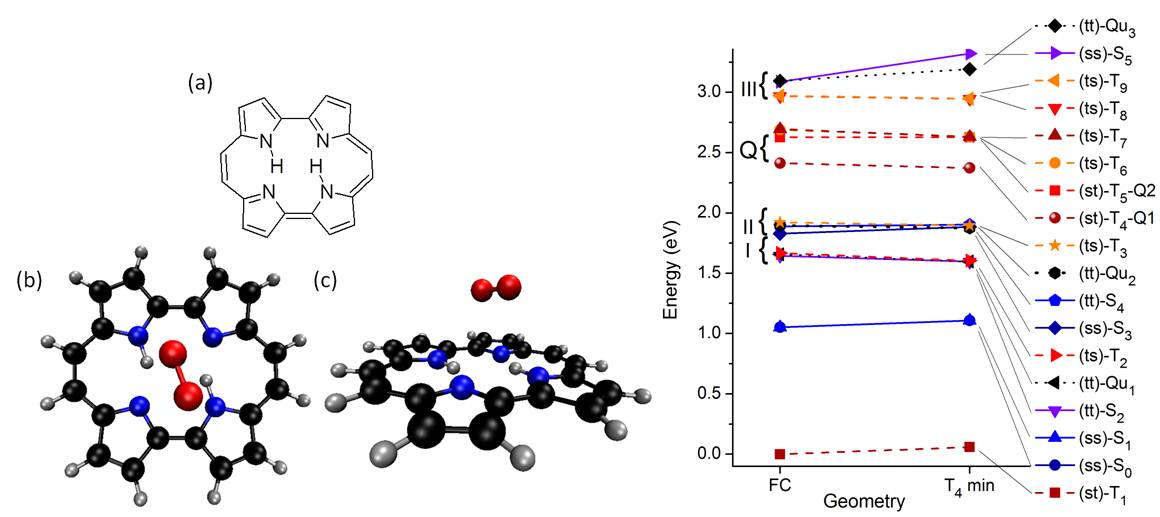
A few years ago, we also investigated porphycenes (PC), a structural isomer of porphine, is a promising PS for photodynamic therapy.
We looked at the electronic ground and excited states of an asymmetric porphycene, 9-amino-2,7,12,17-tetraphenylporphycene (9-ATPPo), using electronic structure calculations (Ref). Different tautomers are considered to address their contributions to the photophysics of 9-ATPPo. Tautomerization pathways on the ground and excited states are constructed between different isomers. It is found that two trans tautomers are mainly responsible for the absorption and emission spectra of 9-ATPPo. These calculations provide a molecular mechanism to explain recent experimental observations, which show a highly complex Q-band structure in the absorption spectrum and pronounced dual fluorescence in the emission spectrum. Furthermore, we show that tautomerization takes place under the assistance of cavity deformations and that a nonradiative process occurs through weak interstate nonadiabatic couplings near the S1 minimum rather than strong ones near conical intersections.
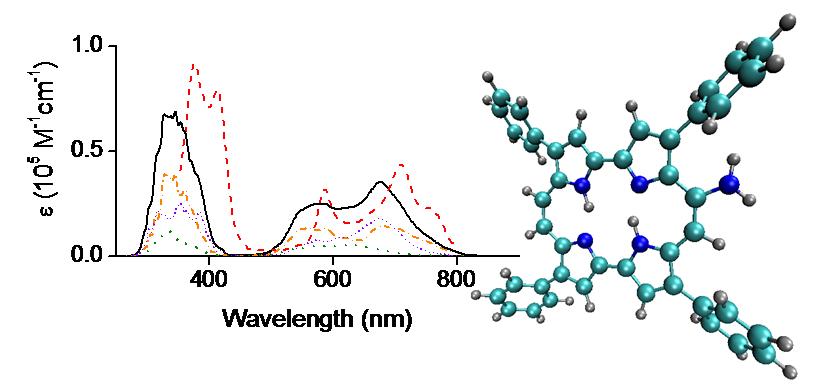
PC excited states can be quenched by molecular oxygen, generating singlet O2. We also investigated the electronic structures of PC and of the PC⋯O2 complex using complete active space perturbation theory (Ref). It is shown that singlet oxygen generation involves 12 electronic states of the complex, with singlet, triplet, and quintet multiplicities. Two scenarios for singlet-O2 yield are analyzed: (I) quenching of triplet states of PC and (II) quenching of singlet states of PC. In the first scenario, which is favored under low O2 concentration, singlet-O2 yield is limited by the relatively low triplet quantum yield of PC. We discussed how the singlet-O2 yield would be busted if conditions for the occurrence of the second scenario could be achieved.