H-bond promotes ring-puckering in adenosine.
In brief:
- After photoexcitation, adenosine conformers with and without intra-molecular hydrogen bonds return to the ground state mainly via ring puckering.
- Internal conversion via H transfer gives a minor contribution.
- Previous experiments assigned the internal conversion to H transfer because they underestimated the excited-state lifetime.
DNA is remarkably stable after absorbing UV thanks to several efficient deactivation processes occurring on time scales ranging from subpicosecond in isolated nucleosides and base pairs to a few hundreds of picoseconds in strands.
In isolated nucleobases, these processes are due to ultrafast internal conversion to the ground state, triggered mainly by conical intersections involving ring puckering. In nucleosides, the presence of the sugar group can open up an additional internal conversion pathway induced by the H-bond between the nucleobase and the sugar.
This additional pathway is the electron-driven proton transfer (EDPT), in which an H-transfer from the sugar to adenine, mediated by a charge-transfer state, creates a conical intersection with the ground state. Experimental results showing a much shorter excited-state lifetime for adenosine than adenine assigned this difference to EDPT.
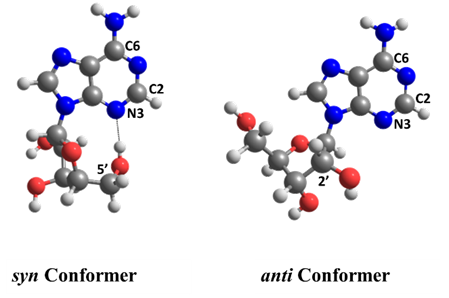
Ritam Mansour, Josene Toldo, and I wanted to quantify how important is EDPT for adenosine’s deactivation in the gas phase. To this goal, we ran surface hopping dynamics for two adenosine conformers with (syn) and without (anti) intramolecular H-bonds.
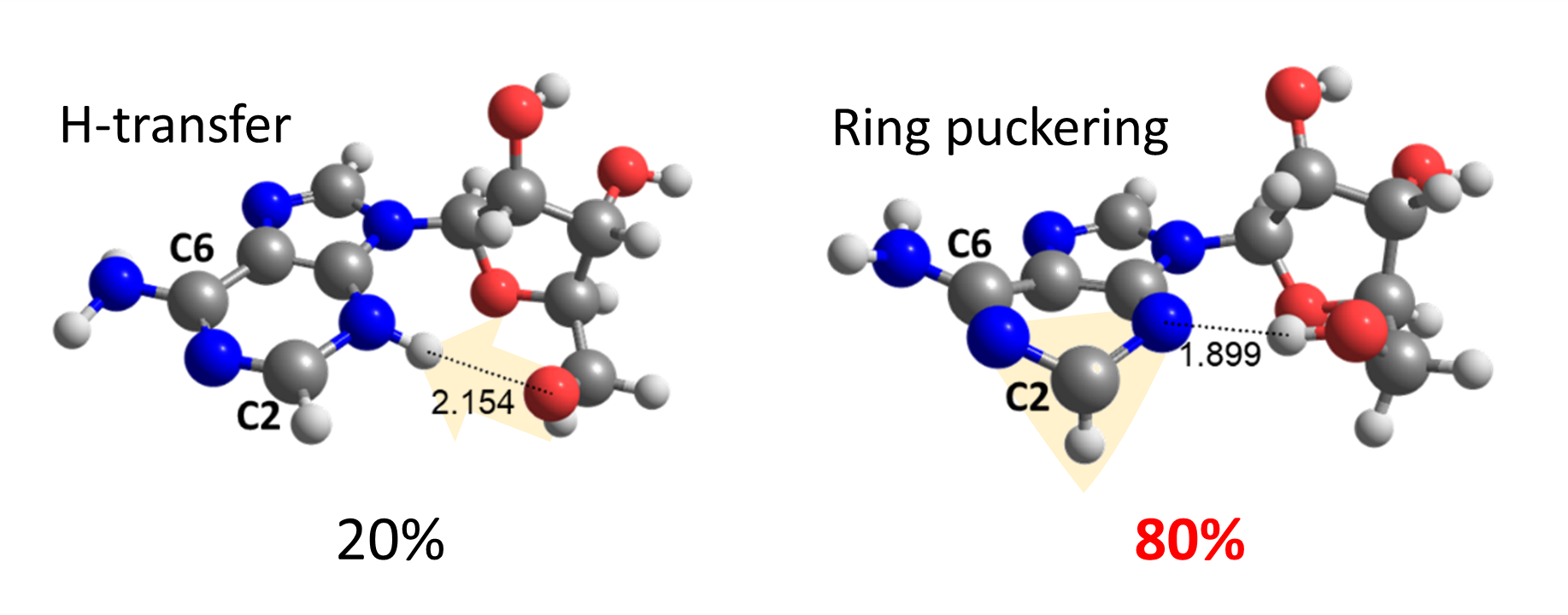
To our surprise, we discovered that the adenine-sugar H-bond is relevant for internal conversion, but not because of EDPT. The H-bond inhibits the relative motion between adenine and sugar, facilitating the ring puckering in adenine.
Thus, ring puckering turns out to answer for 80% of the internal conversion, while H-transfer is responsible for the other 20% only.
These results raise a crucial question: the experiments tell that adenosine’s excited-state lifetime is shorter than that of adenine. But why would it be so if both follow similar puckering pathways?
Our analysis showed that the problem is in the experimental setup. The pump-probe measurements were done, supposing that adenosine’s ionization potential (IP) was constant along the excited-state pathways. However, it’s not. The IP grows so quickly that adenosine leaves the detection window before reaching the S1 minimum.
Thus, the short time constant reported for adenosine did not correspond to the excited-state lifetime.
MB
Reference
[1] R. Mansour, J. M. Toldo, and M. Barbatti. Role of the Hydrogen Bond on the Internal Conversion of Photoexcited Adenosine, J. Phys. Chem. Lett. (2022). DOI: 10.1021/acs.jpclett.2c01554
0 Comments