Dynamics shows that quantum-yield dependence on excitation energy doesn’t occur in the gas phase.
In brief:
- Surface hopping with a semiempirical method is used to simulate the dynamics of trans (E) and cis (Z) urocanic acid.
- Both isomers have ultrafast sub-ps excited-state lifetimes due to E→Z and Z→E processes.
- The Z isomer can alternatively undergo excited-state intramolecular proton transfer.
- No E→Z isomerization quantum-yield dependence on excitation energy is observed in the gas phase.
Urocanic acid (UA) is a UV filter found in human skin, which has been linked to photoimmunosuppression and skin cancer.
After UV excitation, the trans (E) structure of UA can isomerize into cis (Z), while the cis can either turn back to trans or undergo excited-state intramolecular proton transfer (ESIPT).
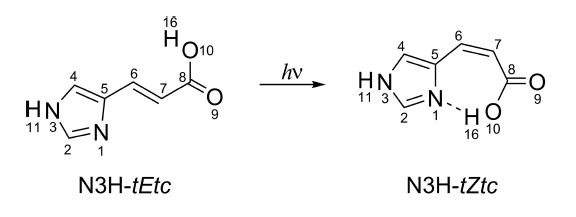
Trans (E)-cis (Z) photoisomerization of urocanic acid (UA).
From the physical-chemical point of view, one of the most puzzling features of UA is in its E→Z photoisomerization quantum yield. Experiments with UA in water showed that when UA is excited at the red tail of its absorption spectrum at 4 eV, the photoisomerization quantum yield is large, about 50%. Nevertheless, by systematically increasing the excitation energy, the quantum yield suddenly drops to about 5% for excitation energies above 4.3 eV.
There are many hypotheses raised to explain this quantum-yield dependence on the excitation energy of E-UA. A few years back, I have proposed that it could be caused by activation of different UA tautomers at different wavelengths. This matter, however, has never been really settled.
In a project led by Deniz Tuna, we have performed the most comprehensive computational simulations of UA in the gas phase so far. We have simulated almost 6000 trajectories, each one for a simulation time of 1.6 ps. The investigation covers the entire conformational space of the E and Z isomers of UA (overall 32 conformers).
UA structure can be generated as a combination of 2 tautomers (A = N1H or N3H), 2 isomers (B = E and Z), and 8 rotamers (x, y, z = c or t), which gives rise to 32 conformers with general structure A-xByz. Click to expand.
These simulations were based on nonadiabatic trajectory-surface-hopping dynamics simulations of photoexcited UA using the semiempirical OM2/MRCI methodology and an adaptive-timestep algorithm. (We had to develop a special procedure to sample initial conditions to account for all those 32 conformers.)
We found out that UA has a sub-ps excited-state lifetime, which is due to ultrafast radiationless excited-state deactivation driven by E↔Z photoisomerization. In the case of Z-UA, Z→E isomerization can be quenched by an excited-state intramolecular proton transfer (ESIPT).
We didn’t find any evidence for an excitation-energy-dependent quantum yield for photoisomerization in E-UA. As you can see in the figure below (left), the quantum yield is constant at about 40% for any excitation energy above the on-set at 4.2 eV.
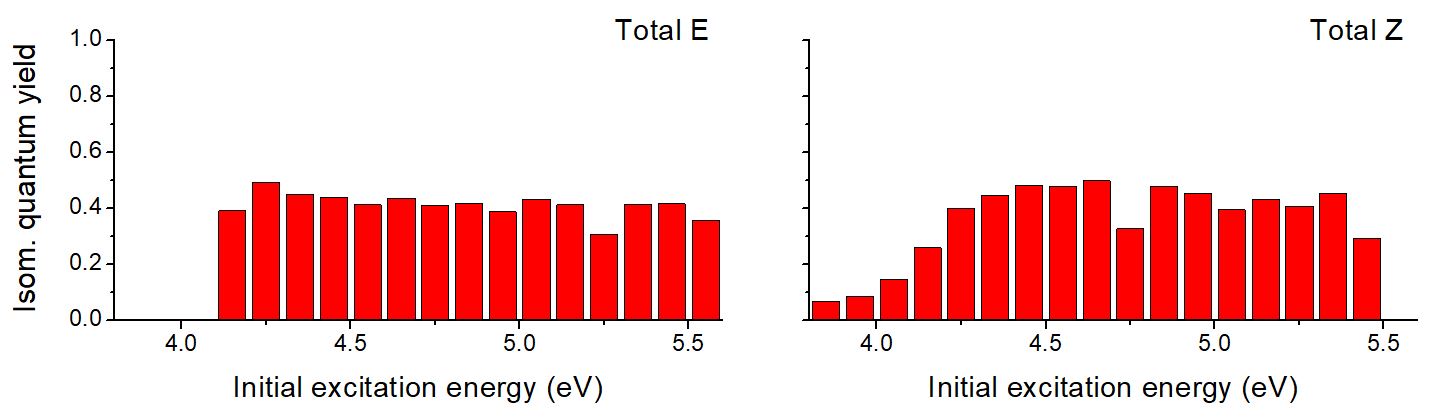
Isomerization quantum yield of E and Z UA.
In the case of Z-UA (right), there the isomerization quantum yield dependence on the excitation energy. It increases from 10 to 40% between 3.8 and 4.3 eV and becomes constant above that. This excitation-energy-dependent quantum yield in Z-UA is caused by competition between photoisomerization and ESIPT processes.
It seems clear that the excitation-energy-dependent quantum-yield of E-UA cannot be explained in the gas phase. It also disproofs my hypothesis that it would be caused by tautomeric effects. At this point, our best guess is that this phenomenon should be caused by the interaction between UA and water in some way.
These results are published in the special issue of Chemical Physics in honor of Wolfgang Domcke [1].
MB
Reference
[1] D, Tuna, L. Spörkel, M. Barbatti, and W. Thiel, Nonadiabatic Dynamics Simulations of Photoexcited Urocanic Acid, Chem. Phys. DOI: 10.1016/j.chemphys.2018.09.036 (2018)