Content
- Dynamics of nucleobases
- Quality of the simulations
- Base models
- Prebiotic synthesis
- Pyrimidine dimers
- Survey of investigations
Dynamics of nucleobases
The five nucleobases occurring naturally in DNA and RNA (ade, gua, cyt, thy, ura) strongly absorb UV radiation. They all are non-fluorescent species, which return to the ground state by internal conversion within a few picoseconds (1 ps = 10-12 s). It is possible that the short lifetime of the nucleobases excited-states is a natural protection of the genetic code against UV induced mutations.
We have investigated how the internal conversion takes place in which of the five nucleobases. These investigations have been carried out with ab initio non-adiabatic dynamics simulations.
The movie below illustrates the internal conversion process for adenine (Ref). After the excitation adenine quickly relaxes in the ππ* excited state. After few hundreds of femtoseconds (1 fs = 10-15 s) the pyrimidine ring undergoes a strong out of plane deformation at the carbon C2. This deformation promotes the conical intersection between the excited and the ground state where adenine deactivates.
In spite of similar deactivation times, each of the five nucleobases follows different pathways. The purine bases adenine and guanine follow homogeneous pathways based on relaxation along the ππ* states. The pyrimidine bases (thymine, cytosine, and uracil) follow inhomogeneous pathways, with sequences of relaxations into local excited-state minima and activation of multiple reaction pathways. These pathways are illustrated in the figure below, which are discussed in details in Ref.
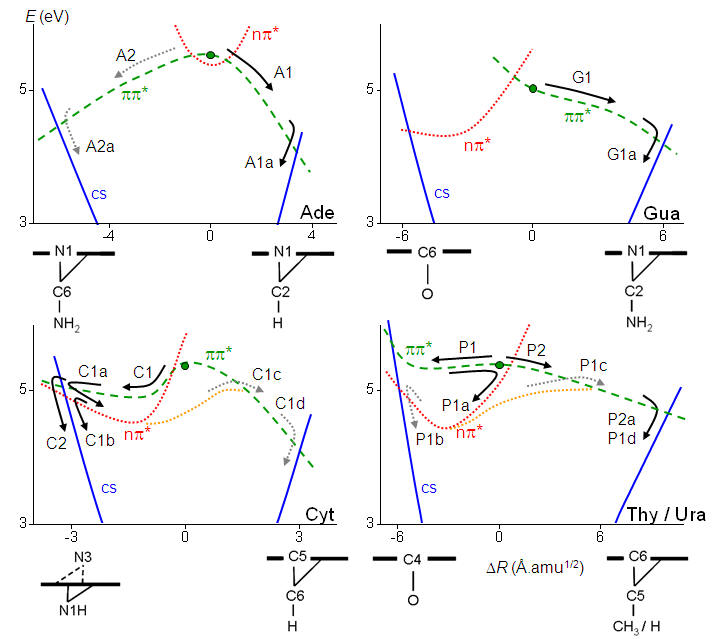
Schematic view of the internal conversion pathways for the five natural nucleobases (Ref).
Quality of the simulations
In spite of the importance of nonadiabatic dynamics simulations for the understanding of ultrafast photo-induced phenomena, simulations based on different methodologies have often led to contradictory results. In Ref, we proceed through a comprehensive investigation of on-the-fly surface-hopping simulations of adenine in the gas phase using different electronic-structure theories (ab initio, semiempirical, and density functional methods, see figure below). Simulations that employ ab initio and semiempirical multireference configuration interaction methods predict the experimentally observed ultrafast deactivation of adenine with similar timescales, however, through different internal conversion channels. Simulations based on time-dependent density functional theory with six different hybrid and range-corrected functionals fail to predict the ultrafast deactivation. The origin of these differences is analyzed by systematic calculations of the relevant reaction pathways, which show that these discrepancies can always be traced back to topographical features of the underlying potential energy surfaces. Later, this benchmark was extended to include ADC(2) (Ref).
![Experimental and theoretical predictions of the ground state population of adenine 1 ps after the photoexcitation.[79]](https://barbatti.org/wp-content/uploads/2014/03/adenine-benchmark.jpg)
Experimental and theoretical predictions of the ground state population of adenine 1 ps after the photoexcitation (Ref).
Base models
To understand the behavior of the nucleobases after UV excitation, it has been useful to simulate not only the nucleobases themselves but also nucleobase models. Molecules like aminopyrimidine and di-aminopyrimidine have been important to allow us to determine the role of the several different reaction pathways and conical intersection available for each nucleobase. For instance, we have recently shown that the stacking interaction typical in nucleic acid strands does not inhibit the out of plane motion which brings the nucleobases to conical intersections. These conclusion where achieved with QM/MM simulations of aminopyrimidine locked between two guanine bases (Ref).
Prebiotic synthesis
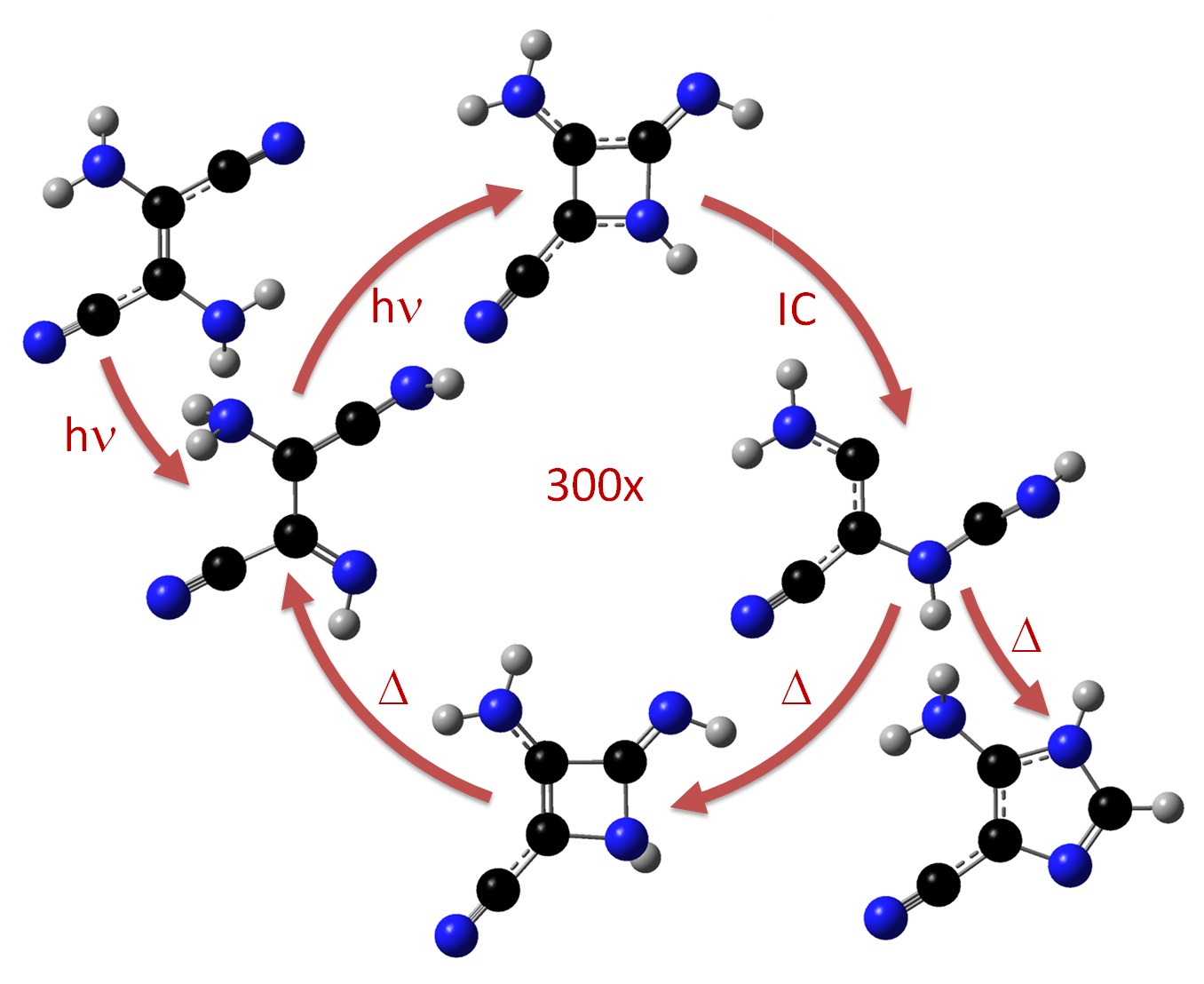
It has been known since the 1960’s that when concentrated hydrogen cyanide (HCN) is irradiated by UV light, it forms an imidazole intermediate that is a key substance for the synthesis of purine nucleobases and nucleotides in abiotic environments. The way how UV radiation acts in this reaction to produce complex organic matter was, however, never clarified. Our simulations have just shown how this process occurs (Ref).
Using diverse computational-chemistry methods, we demonstrated that energy dissipation is too fast to allow the reaction to take place in the hotspot created by the radiation. Alternatively, the reaction proceeds through a series of electronically excited intermediates with low efficiency. Each reactant molecule absorbs hundreds of UV photons before finally gets converted into the imidazole derivative. The researchers also showed that this reaction mechanism may happen in cold environments, as in comets and in terrestrial ices, where spontaneous HCN polymerization is most expected to occur.
The results, which help to understand the role of solar radiation on life’s origin.
Pyrimidine Dimers
Cyclobutane pyrimidine dimers (CPD) are deeply related to mutagenesis and carcinogenesis. It happens when UV radiation induces dimerization of adjacent pyrimidine nucleobases:
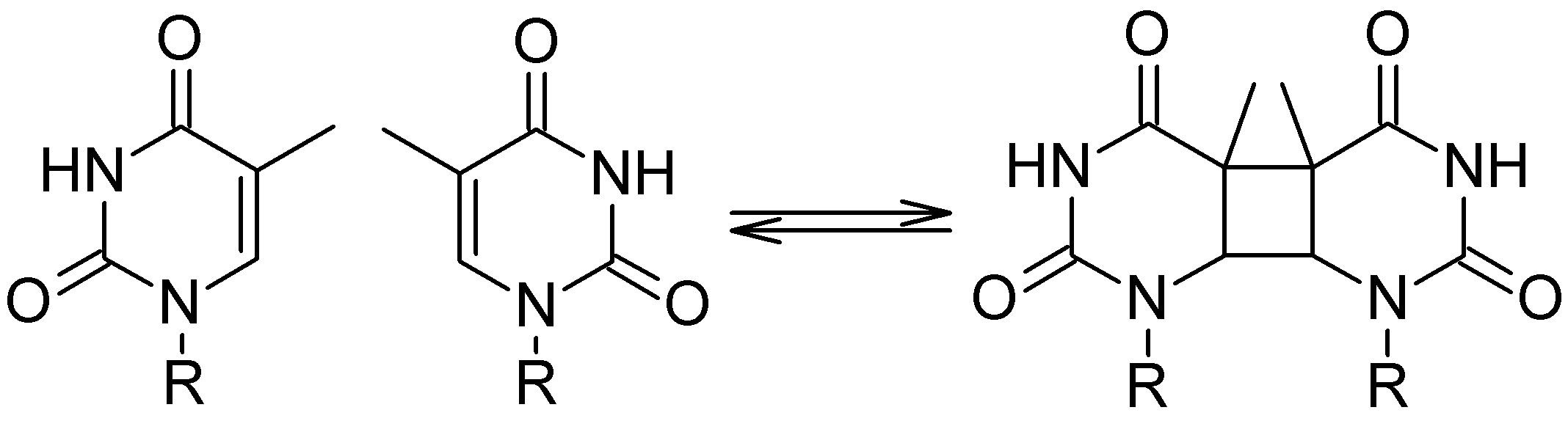
To understand how the dimerization and the repair process happens, I built a benchmark of computational results based on different methods (Ref). For these simulations, I used a thymidine dimer model in the gas phase and I explored the ground and excited potential energy surfaces of neutral (singlet and triplet), cation, and anion species. These surfaces, computed along the two dimerization coordinates C5-C5′ and C6-C6′, look like the figure below.
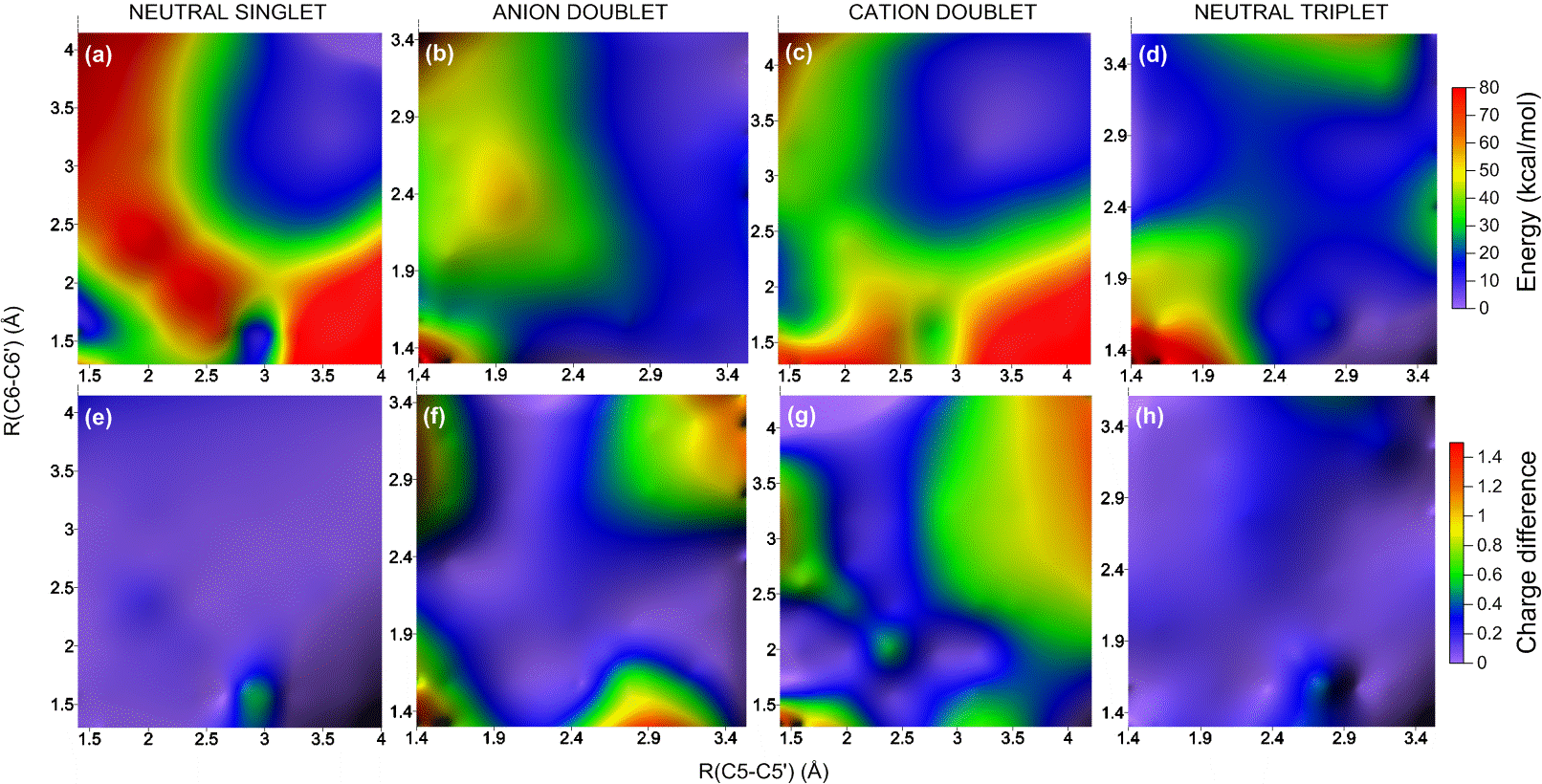
Potential energy surface and charge distribution in the ground state of neutral and charged thymine dimers.
The analysis of such surfaces allowed me to describe several reaction pathways for dimerization and repair, some of them completely unknown so far. For example, the movie below is an animation of one of those paths. It shows the sequential breaking of the dimer on the anion surface.
Important: This video above isn’t dynamics simulation. It is an only linear interpolation between stationary structures (minima and transition states). I built the movie using the time lags measured by Liu et al.
Survey of investigations
| Molecule | Refs. |
| Adenine | Ref Ref Ref Ref Ref Ref |
| Guanine | Ref Ref Ref |
| Cytosine | Ref Ref Ref |
| Thymine | Ref Ref Ref Ref Ref Ref |
| Uracil | Ref Ref Ref Ref |
| Aminopyrimidine | Ref Ref Ref Ref |
| Pyridone | Ref Ref |
| Imidazole | Ref Ref |
| Diaminopyrimidine | Ref Ref |
| Diaminopurine | Ref |
| Trimethylphosphate | Ref |
| Azaindole | Ref Ref Ref |
| Pyrimidine dimers | Ref |
0 Comments