Newton-X 26, a platform for mixed quantum–classical simulations of molecular excited states.
In brief:
-
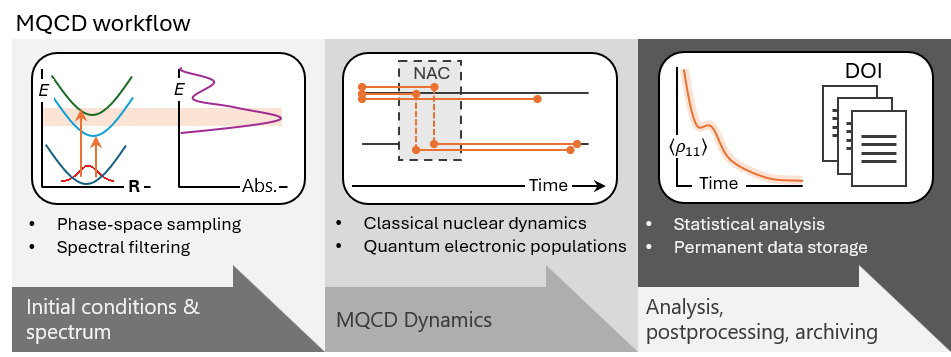
Newton-X 26 covers the complete workflow: spectra, initial conditions, dynamics, analysis, postprocessing, and data archiving.
-
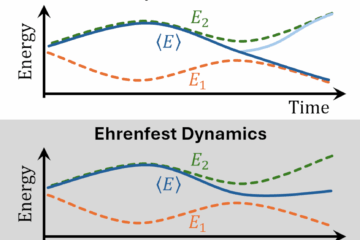
The platform now brings together surface hopping, Ehrenfest dynamics with spontaneous localization, and ab initio multiple spawning.
-
New interfaces, machine-learning acceleration, and FAIR-oriented tools make Newton-X more useful for larger, more diverse, and more reproducible simulations.
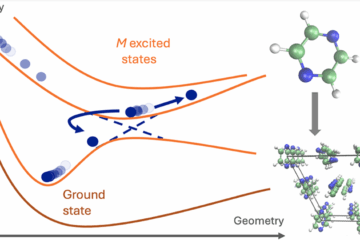
Simulating what happens after a molecule absorbs light is difficult because two kinds of motion become entangled. The nuclei move, roughly like classical particles, while the electrons change their quantum state. These electronic changes may occur in tiny regions of configuration space, where two electronic states come close in energy. That is where nonadiabatic processes often happen.
A full quantum treatment of this coupled electron–nuclear motion is usually too expensive for realistic molecular systems. Mixed quantum–classical dynamics offers a compromise. The nuclei are described by classical trajectories, while the electronic populations evolve quantum mechanically and may transfer between states. This approximation has limitations, but it is one of the main practical routes for simulating nonadiabatic processes in molecular systems.
In this collaborative paper, coordinated from our group and involving a broad Newton-X development community, we present Newton-X 26.
Newton-X has existed for two decades, first as a surface-hopping code and later as a more modular platform for spectra and nonadiabatic dynamics. Newton-X 26 consolidates this evolution into a public, open-source ecosystem.
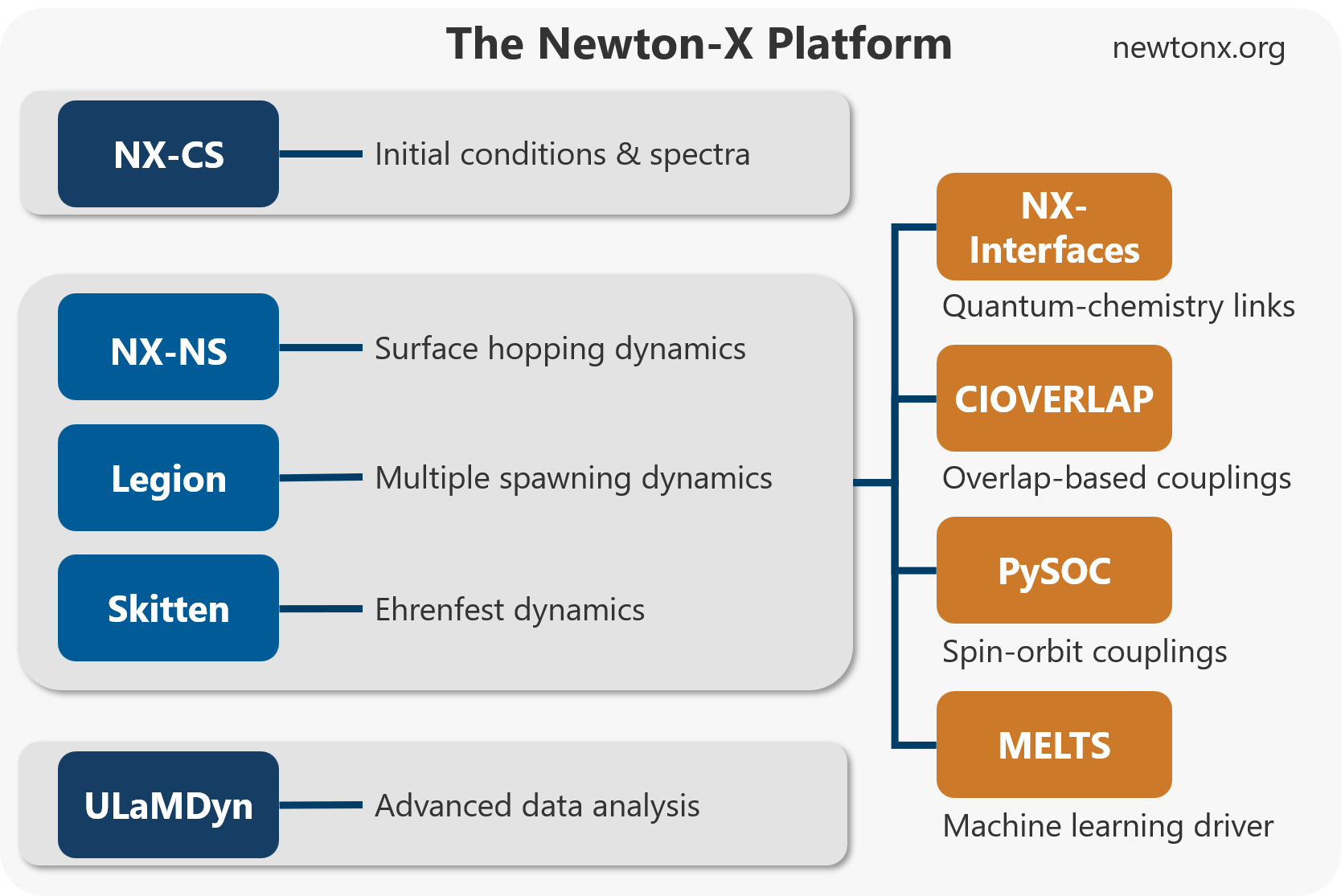
The central idea is simple: a useful simulation platform should enable the several tasks that researchers actually need. Newton-X 26 is organized in workflows that start with spectrum and initial-condition generation, using the NX-CS program. It then propagates mixed quantum–classical dynamics with different engines: NX-NS for surface hopping, Skitten for Ehrenfest dynamics and its spontaneous-localization extension, and Legion for ab initio multiple spawning. After the dynamics, the platform supports analysis, postprocessing, and archiving, including tools such as Ulamdyn for unsupervised analysis of trajectory data.
A major change is methodological breadth. Surface hopping remains the most mature and widely used option in Newton-X. But Newton-X 26 also includes alternatives aimed at different physical regimes. SLED, implemented in Skitten, introduces decoherence into Ehrenfest dynamics through stochastic localization. Legion brings ab initio multiple spawning into the Newton-X ecosystem, giving access to a trajectory-guided wavepacket method when branching and coherence are central.
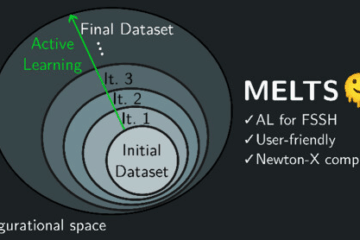
The platform also expands the range of electronic-structure and environment models that can be used. NX-Interfaces connects Newton-X to several quantum-chemistry engines and enable applications ranging from isolated molecules to QM/MM systems, molecular crystals, and periodic systems. Machine-learning-assisted workflows, through MELTS, address another bottleneck: the cost of repeated electronic-structure calculations along many trajectories.
The paper emphasizes something that is easy to neglect: trajectory data are scientific data. They should be curated, analyzed, and archived in reusable forms. This is why Newton-X 26 includes support for postprocessing and FAIR-oriented deposition rather than treating data management as an afterthought.
Newton-X 26 does not remove the hard questions. The reliability of a simulation still depends on the electronic-structure method, the initial ensemble, the dynamical approximation, and the convergence of the trajectory statistics. Some capabilities are more mature than others. That is normal. The contribution of this paper is to provide a clearer, broader, and more sustainable platform on which those choices can be made, tested, and reported.
Cite this paper when using Newton-X 26, describing the current Newton-X software architecture, or referring to its mixed quantum–classical dynamics workflows and interfaces.
MB
Reference
[1] M. Barbatti, R. Souza Mattos, B. Demoulin, M. De Oliveira Bispo, M. Bondanza, M. Brady, R. Crespo-Otero, E. G. De Miranda, P. O. Dral, G. Granucci, A. Hehn, F. J. Hernández, G. Iuzzolino, R. Kar, F. Kossoski, H. Lischka, B. Mennucci, S. Mukherjee, A. Mukhopadhyay, F. Perrella, M. Persico, J. M. Pinheiro, J. Pittner, F. Plasser, N. Rega, E. Sangiogo-Gil, T. Thorat, J. M. Toldo, A. Tomaz, M. T. Varella, L. Vasquez, The Newton-X Platform for Mixed Quantum-Classical Dynamics, Phys. Chem. Chem. Phys. (2026). 10.1039/D6CP01391K