A new version of Dral’s MLatom has been released.
In brief:
- .MLatom 3, an open-source program package, harnesses machine learning (ML) to augment computational chemistry simulations and facilitate the creation of intricate workflows.
- It allows users to execute simulations through command-line options, input files, or Python scripts, accessible on personal computers or the XACS cloud computing service.
- MLatom empowers computational chemists to perform diverse tasks, from calculating energies to simulating spectroscopic phenomena using ML, quantum mechanical, and combined models.
Machine learning (ML) is increasingly becoming a common tool in computational chemistry. At the same time, the rapid development of ML methods requires a flexible software framework for designing custom workflows.
MLatom 3 is a program package designed to leverage the power of ML to enhance typical computational chemistry simulations and to create complex workflows. This open-source package provides plenty of choice to users who can run simulations with the command-line options, input files, or scripts using MLatom as a Python package, both on their computers and on the online XACS cloud computing service at XACScloud.com.
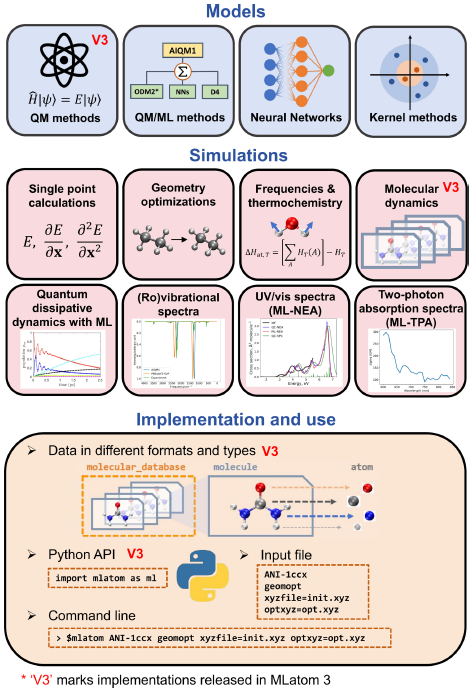
Computational chemists can calculate energies and thermochemical properties, optimize geometries, run molecular and quantum dynamics, and simulate (ro)vibrational, one-photon UV/vis absorption, and two-photon absorption spectra with ML, quantum mechanical, and combined models.
The users can choose from an extensive library of methods containing pre-trained ML models and quantum mechanical approximations such as AIQM1 approaching coupled-cluster accuracy. The developers can build their models using various ML algorithms. The great flexibility of MLatom is mainly due to the extensive use of the interfaces to many state-of-the-art software packages and libraries.
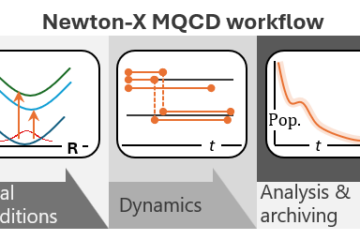
MLatom is mainly designed and developed by Pavlo Dral and his group in Xiamen. Our group in Marseille has contributed to creating the ML-NEA algorithm and helped implement the interfaces to TorchANI, PhysNet, DeePMD-kit, and Newton-X.
MB
Reference
[1] P. O. Dral, F. Ge, Y.-F. Hou, P. Zheng, Y. Chen, M. Barbatti, O. Isayev, C. Wang, B.-X. Xue, M. Pinheiro Jr, Y. Su, Y. Dai, Y. Chen, L. Zhang, S. Zhang, A. Ullah, Q. Zhang, Y. Ou, MLatom 3: A platform for machine learning-enhanced computational chemistry simulations and workflows, J. Chem. Theory Comput. (2024). 10.1021/acs.jctc.3c01203