Unexpected complex isomerization dynamics of ethyl sinapate.
In brief:
- Experiments tracked the isomerization of ethyl sinapate in real time.
- Isomerization quantum yields are strongly dependent on the initially excited isomer.
- Theoretical analysis showed that this dependence is due to a HOOP mode blockage occurring only in one isomer when leaving the conical intersection.
Sinapate esters have been extensively studied for their potential application in nature-inspired sunscreen formulations and photothermal materials. They efficiently absorb UV radiation and dissipates it as heat after internal conversion.
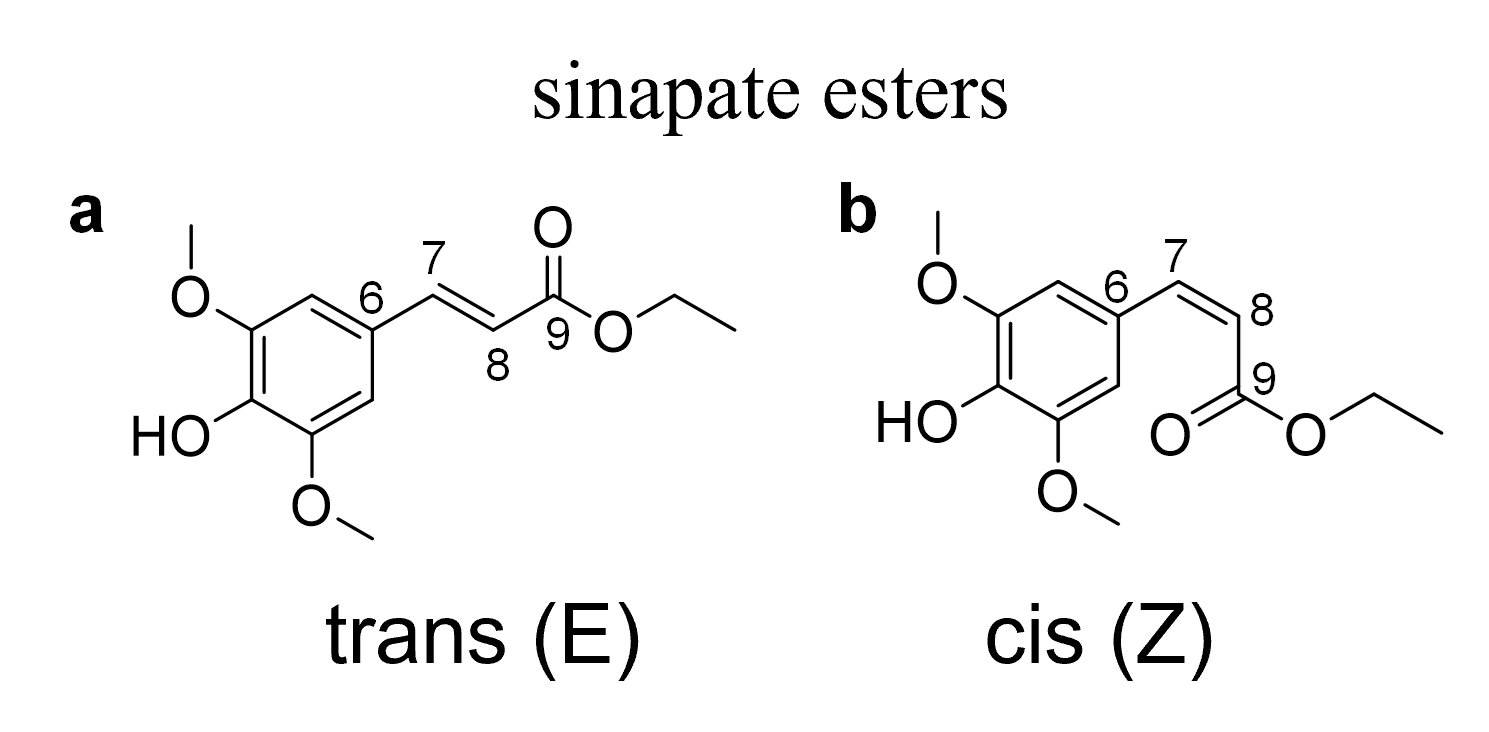
Trans and cis structures of ethyl sinapate (ES).
There is general consensus that the relaxation mechanism of sinapate esters following photoexcitation is mediated by geometric isomerization between cis and trans isomers.
This relaxation has been largely inferred through indirect studies involving transient electronic absorption spectroscopy in conjunction with steady-state spectroscopies. However, to date, there is no direct experimental evidence tracking the formation of the photoisomer in real-time.
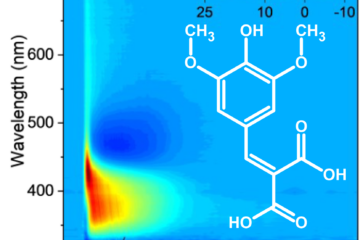
Temitope T. Abiola, from Vas Stravos’ group in Warwick, used transient vibrational absorption spectroscopy to measure direct structural changes occurring in ethyl sinapate (ES) after UV excitation.
As expected, torsional isomerization dominates the internal conversion. The experimental signals clearly distinguish the repopulation of the starting isomer in the S0, isomer photoproduct formation, and vibrational cooling in the electronic ground state.
However, the quantum yields and time constants show some non-trivial features:
- Starting from trans-ES, the probability of getting either trans or cis photoproduct is about 1/2.
- Starting from cis-ES, the probability of recovering cis is 3/4, and forming trans is 1/4.
Moreover, no matter the initial isomer, recovering the original isomer is at least twice faster as getting the other isomer.
Thus, sinapate ester’s geometric isomerization is surprisingly more complex than originally thought.
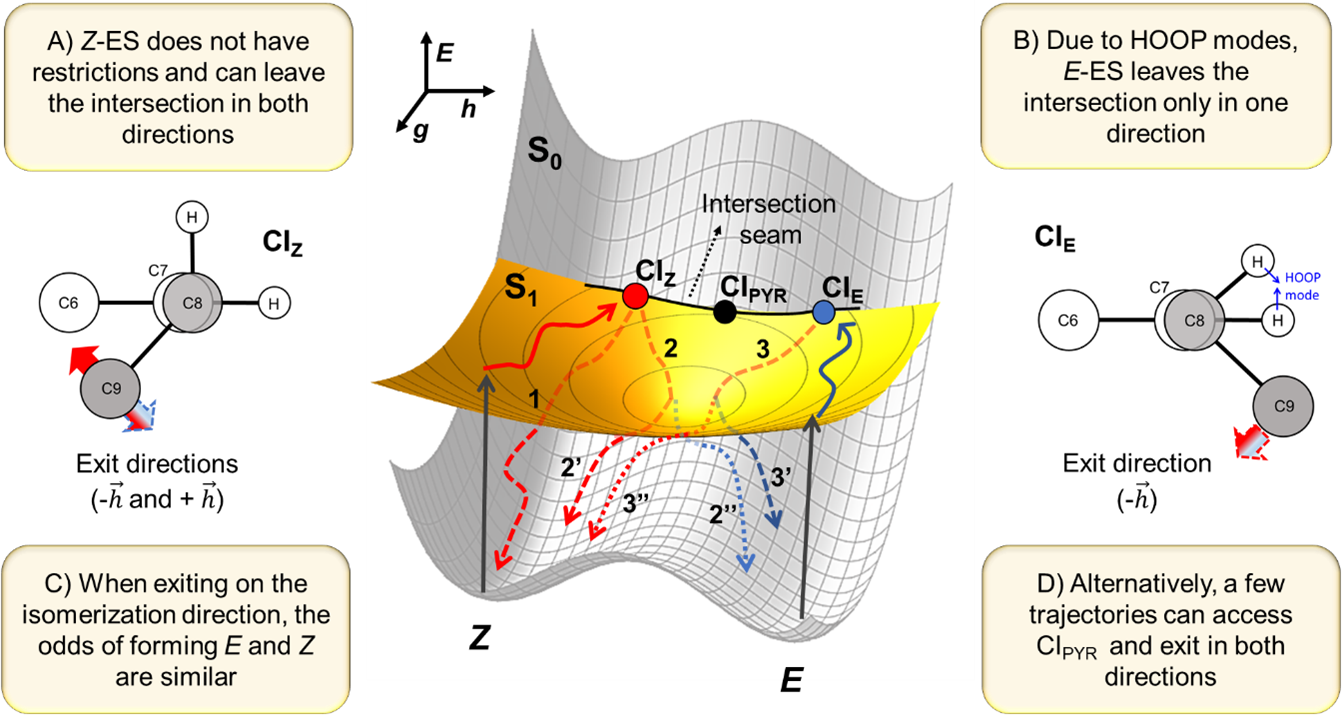
Schematic representation of the conical intersection geometries and PES for the photoisomerisation starting from cis (Z) and trans (E) isomers. Solid red and blue lines indicate relaxation on the S1 surface while dashed lines indicate relaxation in the ground state.
Computational chemistry, under Josene Toldo’s leadership, was instrumental to shed light on the mechanism driving such isomerization.
We computed the intersection seam between the excited and the ground state where the molecules could do the internal conversion. Then, we analyzed the reaction pathways starting from the Franck-Condon region till the crossing seam.
We learned that ES tends to find the point of the crossing seam that still has the initial isomeric structure. Thus, it’s clear why recovering the original isomer is faster: cis-ES converts to the ground state at a cis-conical intersection. Trans-ES do so near a trans-conical intersection. Therefore, It always takes longer to form the other isomer.
But why are the quantum yields dependent on the initial isomer?
The answer to this question is more involved. We must look at the branching space directions defining the intersection seam.
When cis-ES is excited, it reaches a cis intersection. From there, it can leave the intersection twisting back to cis (and likely recovering cis). It can also leave the intersection twisting in the trans direction. In this case, it can either continue and form the trans product or stop the twist and return to cis. Thus, after exciting cis-ES, we end up with roughly 3/4 of cis and 1/4 of trans.
On the other hand, when trans-ES is excited, it reaches a trans intersection. From there, ES cannot leave the intersection twisting back to trans (recovering trans) because this motion is blocked by a HOOP (hydrogen ou-of-plane) mode. ES can only leave the trans-intersection twisting in the cis direction. Then, it can either form the cis product or return to trans, yielding 1/2 of cis and 1/2 of trans products.
MB
Reference
[1] T. T. Abiola, J. M. Toldo, M. T. do Casal, A. L. Flourat, B. Rioux, J. M. Woolley, D. Murdock, F. Allais, M. Barbatti, V. G. Stavros, Direct structural observation of ultrafast photoisomerization dynamics in sinapate esters, Commun. Chem. 5, 141 (2022). DOI: 10.1038/s42004-022-00757-6
0 Comments