New method enables nonadiabatic dynamics based on energy-gaps’ time-derivatives.
In brief:
- We proposed a novel model for computing approximated nonadiabatic couplings, named time-dependent Baeck-An (TD-BA).
- In TD-BA, couplings are a function of the energy gaps and their second time-derivatives. TD-BA be directly employed in fewest-switches surface hopping.
- We implemented TD-BA into Newton-X and tested it on ethylene and fulvene dynamics, with excellent results.
Surface hopping has become one of the main tools for nonadiabatic dynamics simulations of photoexcited molecules. However, the primary constraint for interfacing surface hopping to a new quantum chemical method or program is to get the first-order nonadiabatic coupling vectors
{{\mathbf{h}}_{JL}}=\left\langle J | \tfrac{\partial }{\partial \mathbf{R}}L \right\rangleIn this work, we present a new approach to compute these couplings.
In the Baeck-An (BA) approximation, first-order nonadiabatic coupling vectors are given in terms of adiabatic energy gaps and the second derivative of the gaps with respect to the coupling coordinate.
We derived a time-dependent (TD) BA approximation, where the couplings (projected on the nuclear coordinates v) are computed from the energy gaps and their second time-derivatives:
\mathbf{v}\cdot {{\mathbf{h}}_{JL}}=\frac{1}{2}\sqrt{\frac{1}{\Delta {{E}_{JL}}}\frac{{{d}^{2}}\Delta {{E}_{JL}}}{d{{t}^{2}}}}TD-BA couplings can be directly used in the fewest switches surface hopping (FSSH) approach, where hopping probabilities are proportional to \mathbf{v}\cdot {{\mathbf{h}}_{JL}}. Thus, TD-BA enables nonadiabatic dynamics with any electronic structure methods able to provide excitation energies and energy gradients.
Test results of surface hopping with TD-BA couplings for ethylene and fulvene show that the TD-BA approximation delivers a qualitatively correct picture of the dynamics and a semiquantitative agreement with reference data computed with exact couplings. Nevertheless, TD-BA does not perform well in situations conjugating strong couplings and small velocities.
Considered the uncertainties in the method, TD-BA couplings could be a competitive approach for inexpensive, exploratory dynamics with a small trajectories ensemble.
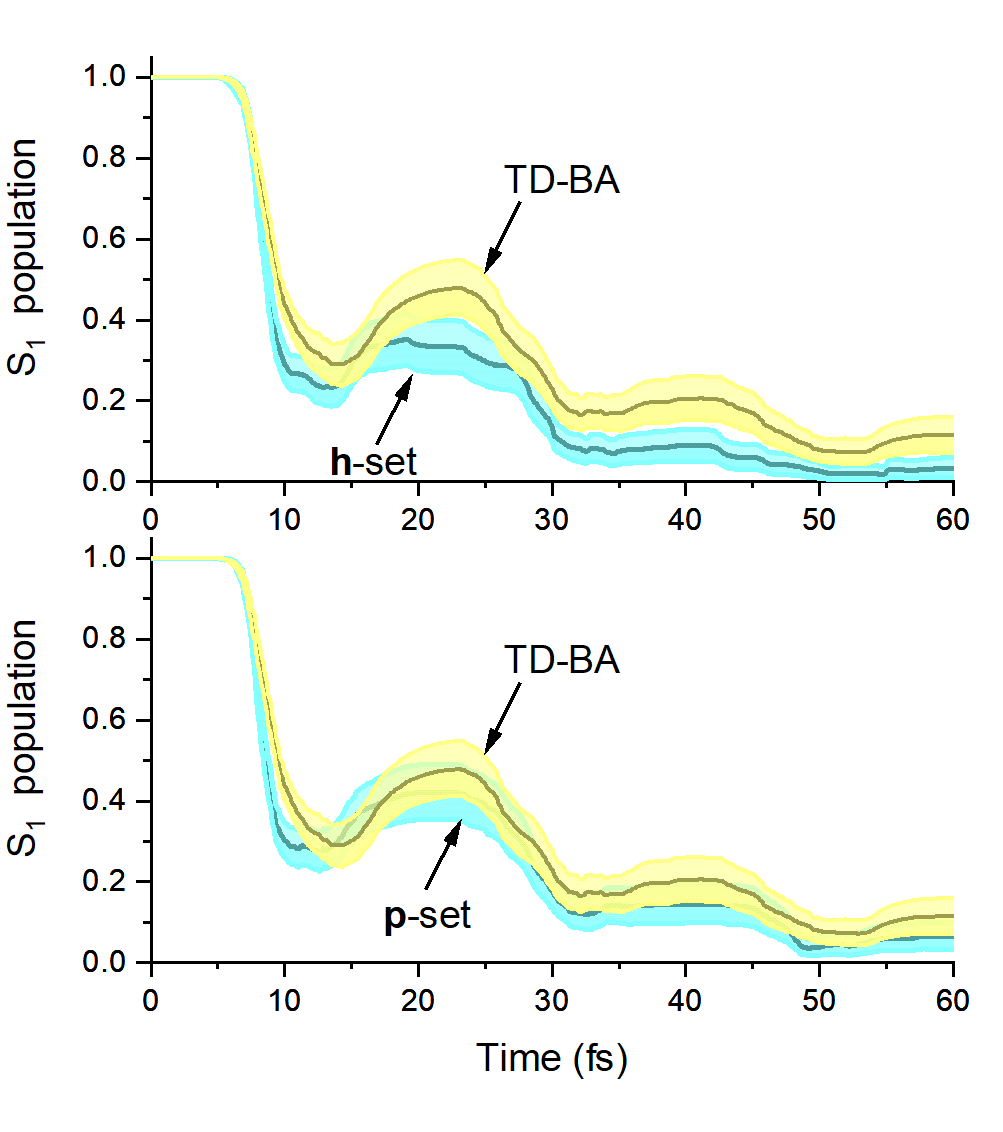
Excited-state population of fulvene as a function of time computed with TD-BA. It is compared to FSSH dynamics with exact couplings, with velocity after hopping adjusted along the coupling (h-set) and the momentum (p-set) directions.
We also assessed the potential use of TD-BA couplings for surface hopping dynamics with time-dependent density functional theory (TDDFT), but the results are not encouraging due to singlet instabilities near the crossing seam with the ground state.
The TD-BA model is not supposed to replace the exact calculation of nonadiabatic couplings in surface hopping. It can be, however, an alternative approach to be used with electronic structure methods for which nonadiabatic couplings are still unavailable or are too computationally expensive to be computed on the fly. The TD-BA model can also be used as an inexpensive diagnostic of nonadiabatic interactions during exploratory dynamics, triggering higher-level calculations.
FSSH based on TD-BA couplings is available in Newton-X version 2.2 build 15 or higher, to be used with any of the electronic structure methods and programs interfaced to Newton-X.
MB
Reference
[1] M. T. do Casal, J. Toldo, M. Pinheiro Jr, and M. Barbatti, Fewest switches surface hopping with Baeck-An couplings [version 1; peer review: 2 approved], Open Res. Europe 1, 49 (2021). https://doi.org/10.12688/openreseurope.13624.1