Machine learning enables high-precision spectrum simulations.
In brief:
- Machine Learning (ML) can be used to increase the precision of the nuclear ensemble approach (NEA).
- The ML-NEA approach is based on kernel ridge regression with a global descriptor (KREG).
- ML-NEA allows statistically converged NEA simulations employing only a few hundreds of quantum chemical calculations.
- With ML-NEA, there is no need to adjust the bandwidth parameter.
In collaboration with Pavlo Dral’s group, we developed a new ML approach to compute absorption cross-sections within the nuclear ensemble approach (NEA).
In the ML-NEA approach, a large ensemble with 50,000 nuclear geometries is obtained by stochastically sampling a Wigner distribution for the quantum harmonic oscillator. The required electronic properties — excitation energies and oscillator strengths — are calculated only for a fraction of these points using the reference electronic structure method (here, we used linear-response TDDFT).
The obtained data set is used as the training set for ML. We train individual ML models for each of the reference properties using the KREG model (KRR with the Gaussian kernel function and the RE descriptor) as implemented in MLatom to calculate the required properties for the remaining points in the large ensemble. The combined set of reference and ML properties is used to calculate NEA cross-section as implemented in Newton-X.
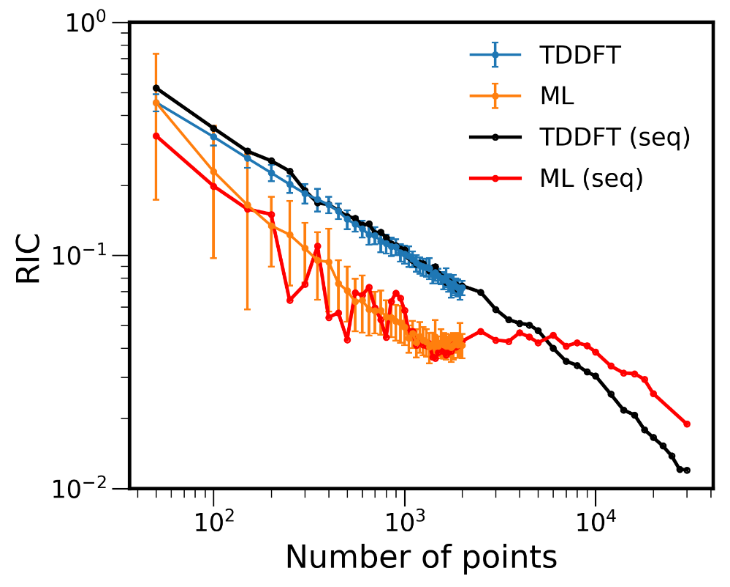
The graph shows the errors between ML-NEA and TDDFT-NEA cross-sections relative to a benchmark TDDFT-NEA calculation. For any number of training points smaller than 5000, ML-NEA (orange) is more accurate than pure TDDFT NEA (blue) computed with an ensemble of the same size.
We also suggest using the ML validation-set errors to gauge the convergence of the ML-NEA model. It provides a criterium for defining the required number of reference data calculated with electronic structure methods. Such a criterium is convenient for calculating NEA cross-sections of new molecules when it is not clear how many electronic structure calculations should be performed to obtain satisfactory accuracy and precision.
ML-NEA allows refining cross-sections calculated earlier with relatively small ensembles without incurring substantial additional cost.
It brings the following benefits:
- ML-NEA allows obtaining high precision and high accuracy NEA cross-sections for small to medium-size molecules at the cost of a few hundreds of single-point QC calculations. To achieve the same quality of pure QC-NEA cross-sections would require tens of thousands of QC calculations.
- ML-NEA eliminates the need to use the line-shape arbitrary broadening parameter δ to smooth the NEA cross-section. This parameter is fixed to a tiny constant value that does not impact the final result.
We anticipate that ML-NEA may routinely allow obtaining NEA absorption cross-sections for medium to large size molecules with reasonable accuracy and high precision at the cost of about one hundred QC single points. It may also allow sampling regions of the configurational space with a low population at the cost of a few hundred QC calculations.
Although we tested ML-NEA for absorption spectra based on Wigner distributions and TDDFT electronic structure, the method is much more general. It can be used for other types of NEA spectra, and any quantum chemical method able to compute excitation energies and transition moments.
A step-by-step tutorial on how to use ML-NEA is available here.
MB
Reference
[1] B.-X. Xue, M. Barbatti, P. O. Dral, Machine Learning for Absorption Cross Sections, J. Phys. Chem. A. DOI:10.1021/acs.jpca.0c05310 (2020).