Paper reviews the last ten years of developments in nonadiabatic dynamics.
In brief:
- The main and most recent developments in nonadiabatic mixed quantum-classical dynamics are presented.
- Electronic structure methods are reviewed in the context of nonadiabatic dynamics.
- Software and other computational aspects are critically assessed.
It’s a pleasure to announce the publication of Recent Advances and Perspectives on Nonadiabatic Mixed Quantum-Classical Dynamics in the Chemical Reviews [1]. In this work, Rachel Crespo-Otero an I:
- Describe the standard methods for nonadiabatic mixed quantum-classical (NA-MQC) dynamics.
- Discuss the NA-MQC methods developed within the last decade.
- Analyse the main electronic structure methods used with NA-MQC dynamics.
- Present methods for spectroscopic simulations based on NA-MQC dynamics.
- Survey the main software to run NA-MQC simulations.
- Critically assess the accuracy and precision of NA-MQC dynamics.
NA-MQC dynamics has become one of the central tools in quantum chemistry to investigated ultrafast excited-state phenomena. Fed by an on-the-fly link to electronic structure methods, NA-MQC dynamics prescind of pre-computed multidimensional potential energy surfaces, allowing simulations of large and realistic molecular systems.
The core approximation in NA-MQC dynamics is a split between degrees-of-freedom that are treated classically (usually, nuclei) and degrees-of-freedom that are treated quantum-mechanically (usually, electrons). The self-consistency between these two domains is ensured by an algorithm that transfers information between them.
Nonadiabatic effects — the interaction between nuclei and electrons beyond the Born-Oppenheimer approximation — are introduced in different ways in NA-MQC dynamics; the most common ones being by
- averaging states,
- hopping between states, or
- spawning new basis functions to other states.
Such flexibility gives rise to many different methods, among which the mean-field Ehrenfest dynamics, surface hopping, and multiple spawning are the most well known.
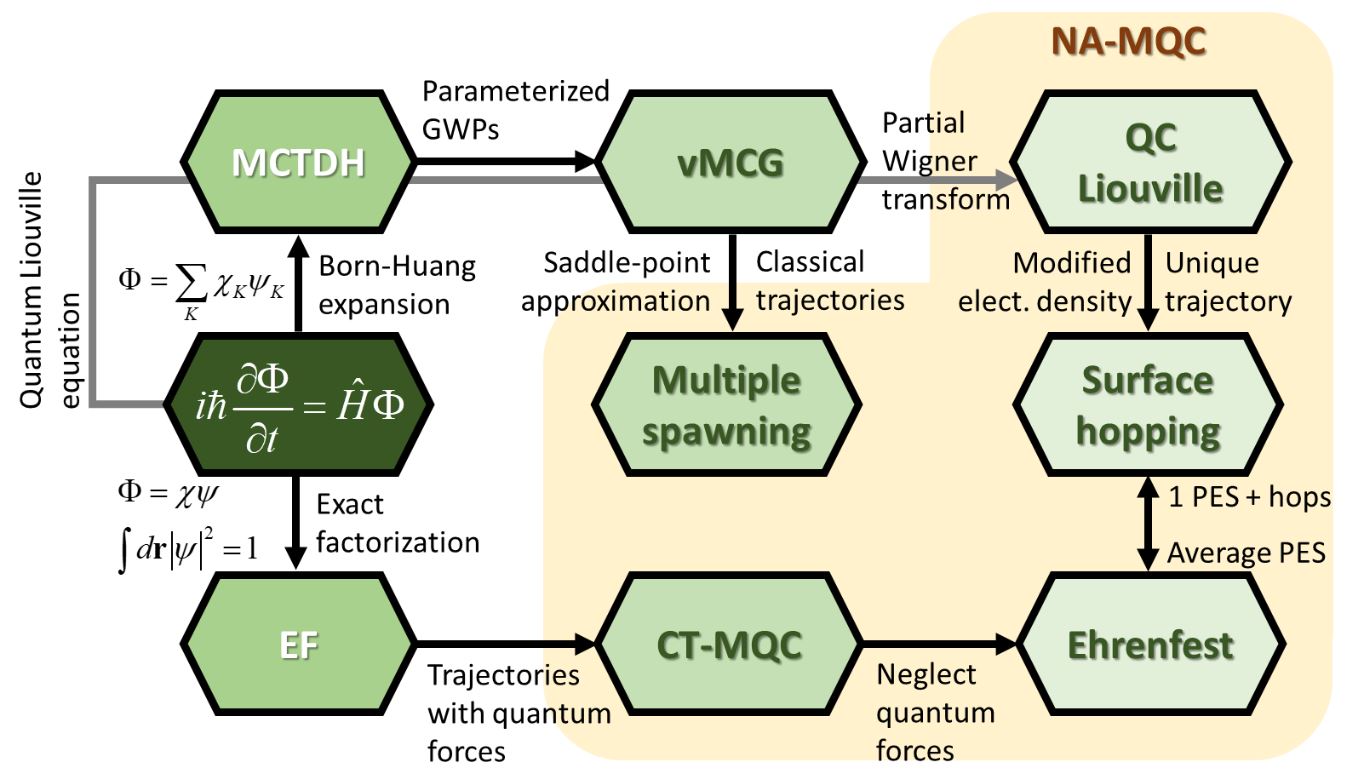
There are different ways to solve the time-dependent Schrödinger equation for molecules in the nonadiabatic regime. All of them may be cast into a mixed quantum-classical approximation (NA-MQC).
In the last decade, the number of new developments in NA-MQC dynamics has immensely grown, driven by the advances in computational capabilities and new electronic structure methods. The pace of these developments is such that it’s getting truly hard to keep track of them. Rachel and I have surveyed about 500 papers to build a panorama of the main advances and present them in a systematic way.
We also discuss the electronic structure methods for excited states used in the context of NA-MQC dynamics, from ab initio multireference wavefunction theory to semiempirical single-reference density functional theory.
For people interested in applying NA-MQC dynamics, the review provides practical information on the main software available today. (Including Newton-X, naturally.)
Accuracy and precision of NA-MQC dynamics simulations are major issues that, if not adequately addressed, may lead to qualitatively wrong results. In the review, we discuss the main source of problems and deliver a guide for best practice.
NA-MQC dynamics is a vast research field and we reached out to the community to get their feedback on our review. Rachel and I are grateful for discussions with N. Ferré, M. Filatov, E. Fromager, G. Groenhof, M. Huix-Rotllant, R. Lindh, and J. W. Park. We are also in debt to A. Akimov, B. Churchod, M. Dommett, L. González, G. Granucci, S. Mai, F. Plasser, J. Subotnik, R. Szabla, I. Tavernelli, S. Tretiak, and O. Weingart for their comments on the manuscript.
MB
Reference
[1] R. Crespo-Otero and M. Barbatti, Recent Advances and Perspectives on Nonadiabatic Mixed Quantum-Classical Dynamics, Chem. Rev. doi: 10.1021/acs.chemrev.7b00577 (2018).
1 Comment
Dynamics of UV-Excited Thymine-Water Cluster – Light and Molecules · August 12, 2018 at 2:53 PM
[…] Dynamics was performed with decoherence-corrected surface hopping based on ADC(2) electronic structure. […]
Comments are closed.