A rigid, stacked aromatic scaffold that relaxes fast, then stubbornly refuses to leave the excited state.
In brief:
-
Ultrafast funnel: photoexcitation into a weak S₃ shoulder relaxes to S₁ in ~0.1 ps via S₂.
-
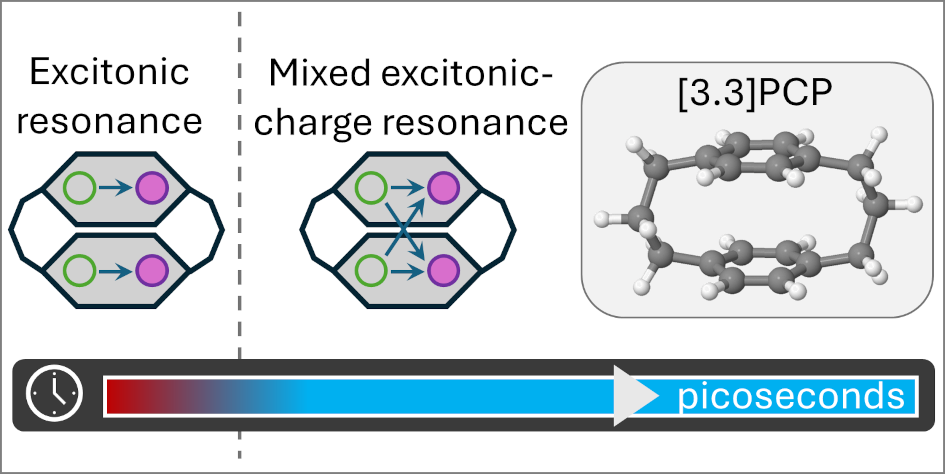
Electronic “identity shift”: the excited state evolves from mostly excitonic to a mixed excitonic/charge-resonance regime (CT rising to ~0.5).
-
A structural driver: π-stack contraction and an inter-ring breathing mode dominate the picosecond dynamics (≈172 cm⁻¹ in the trajectories).
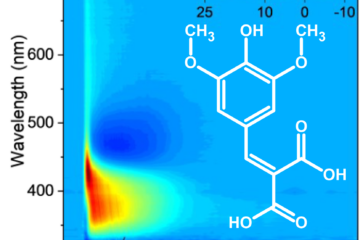
In a project that was the core of Muhammad Tahir Hafeez’s Master’s thesis, we examined what happens when [3.3]paracyclophane ([3.3]PCP) absorbs ultraviolet light and is promoted to its electronically excited manifold. Paracyclophanes are peculiar molecules: two benzene rings are forced into a parallel, face-to-face arrangement by short aliphatic bridges. That enforced geometry makes through-space π–π interactions unavoidable, and those interactions can create excimer-like electronic states that are both chemically interesting and methodologically annoying (in a productive way).
We tackled the problem with time-dependent DFT (TDDFT) combined with Fewest Switches Surface Hopping (FSSH). FSSH is the mixed quantum-classic workhorse introduced by Tully and, as usual, we used a decoherence corrections to keep the electronic amplitudes from remaining unrealistically coherent for too long. Those choices matter, because [3.3]PCP is exactly the kind of system that exposes the sharp edges of approximate nonadiabatic dynamics.
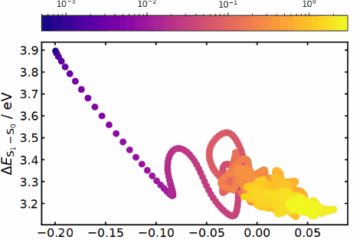
The initial excitation is not into the brightest band. Instead, we select initial conditions in the low-energy shoulder associated with S₃, and we see an ultrafast cascade: S₃ depopulates in roughly 50 fs, population briefly visits S₂, and then the molecule lands in S₁, where it becomes kinetically trapped on the 2-ps simulation window. In other words: fast relaxation down, slow escape out.
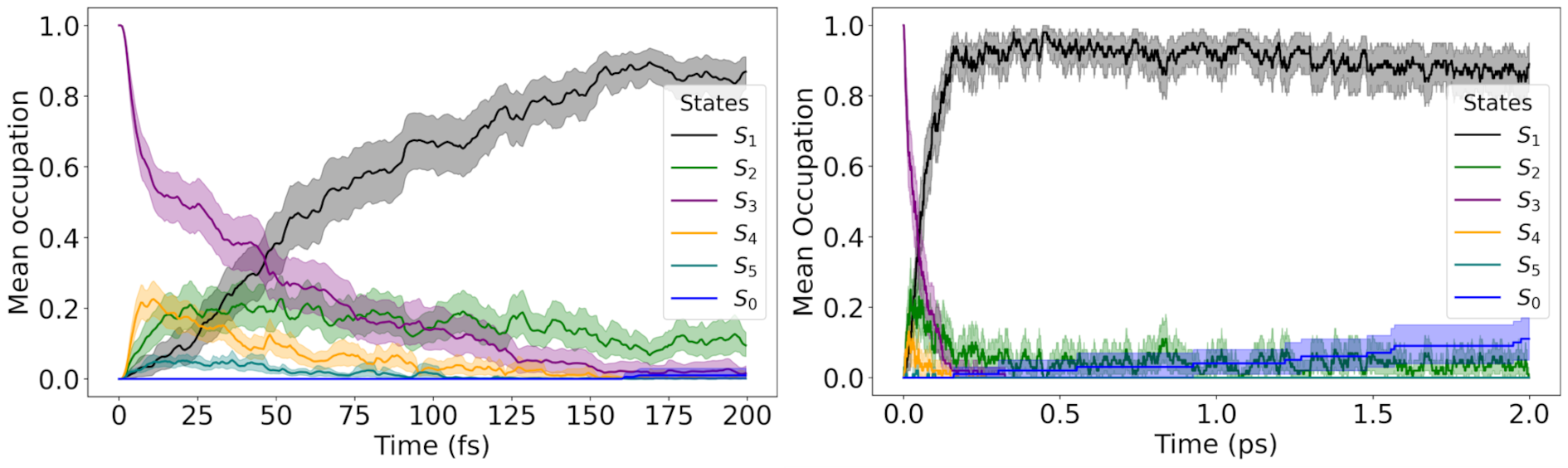
What is this trapped S₁ state, electronically speaking? To answer that, we used a fragment-based analysis of the one-electron transition density matrix and monitored two compact descriptors: a charge-transfer (CT) measure and a participation ratio (PR) for delocalization. The picture is clean. Early on, the excitation has mainly excitonic resonance character, but within the first ~200 fs it morphs into a long-lived mixed excitonic/charge-resonance regime, with CT stabilizing near ~0.5 and PR indicating delocalization over both rings (and some participation of the bridges, depending on the partition).
The nuclear motion that accompanies this electronic reorganization is equally telling. The inter-ring distance contracts rapidly (from the ground-state value around 3.25 Å toward the S₁ minimum near 2.94 Å) and then oscillates in an inter-ring breathing mode that dominates the subsequent dynamics. This connects nicely to recent ultrafast work on [2.2]PCP, which also highlighted a breathing coordinate modulating excimer features—suggesting that these cyclophanes share a common “excimer-forming” structural pathway.
Finally, we estimate that S₁→S₀ recovery is slow (effective lifetime on the order of tens of picoseconds within our model and criteria), largely because reaching the relevant S₁/S₀ crossing region is energetically and geometrically constrained. We also make the obvious caution explicit: these numbers are method-dependent, and condensed-phase dissipation would likely change the long-time behavior.
MB
Reference
[1] M. T. Hafeez, R. S. Mattos, L. Ibele, M. Barbatti, Ultrafast Dynamics and Excited-State Trapping in [3.3]Paracyclophane, Phys. Chem. Chem. Phys. (2026). 10.1039/D5CP04083C