How a smart feature and a pinch of phase-fixing let machine learning predict nonadiabatic couplings like a pro.
In brief:
-
Direct ML prediction of nonadiabatic coupling (NAC) vectors now works reliably.
-
The winning recipe: use the gradient difference between states as the descriptor and fix random sign flips.
-
ML-driven surface hopping matches trusted quantum benchmarks while running hundreds of times faster.
If you’ve ever tried to simulate photochemistry, you’ve met the troublemaker of the story: the nonadiabatic coupling vector, or NAC. It tells you how fast a molecule can jump between electronic states as the nuclei move. The catch? NACs are touchy. They flip sign with minor tweaks, spike near conical intersections, and are notoriously expensive to compute.
For years, machine learning tried to learn NACs straight from geometry—and kept tripping. The new twist, developed by Jakub Martinka, is disarmingly simple: don’t make the model guess the physics—feed it the physics. The key input is the gradient difference between the two electronic states (call it Δ∇E). This vector already points in the direction of the privileged state interaction. In other words, Δ∇E carries the part of nonadiabatic information, so the model doesn’t have to discover it from scratch.
There’s a second trick. NACs have an arbitrary sign (a phase) that can flip from one geometry to the next, even when nothing physical changes. If you train on raw data, the model learns noise. We added a phase-correction loop: try both signs during training and keep the choice that improves the fit.
Put together, these two moves—physics-informed descriptor + phase correction—turn the NAC problem from slippery to solid. In tests on classic photochemical playgrounds (fulvene and the protonated methanimine CH₂NH₂⁺), the machine-learned NACs reach near-perfect correlation with the quantum-chemistry references. When you plug these learned NACs into the fewest-switches surface hopping (FSSH), the population dynamics match the benchmarks. The ML engine reproduces when and how often hops occur, which states they connect, and how the excited state decays in time.
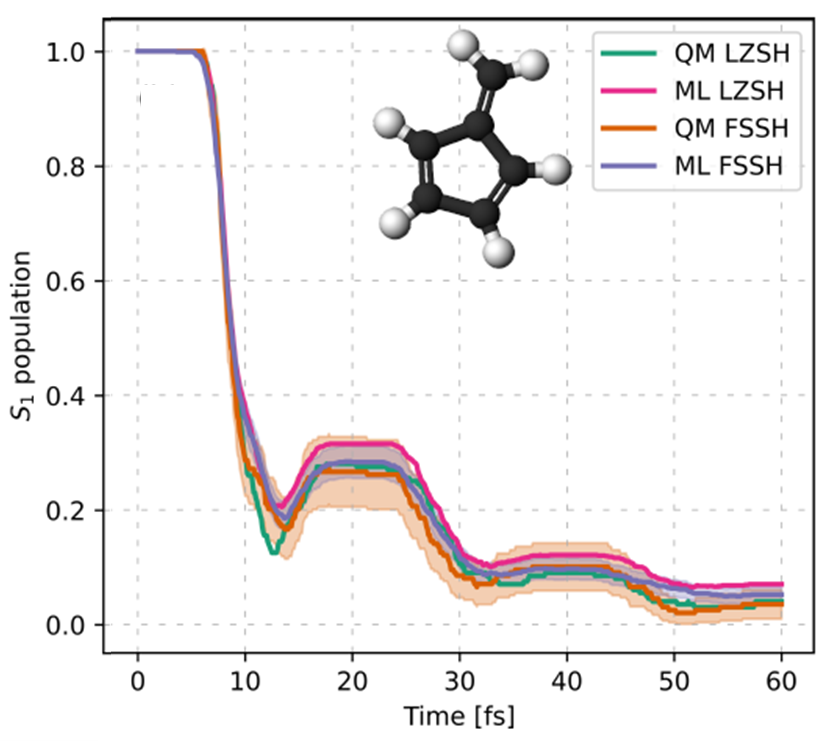
Excited-state population of fulvene as a function of time computed with ML and QM NAC vectors.
Now the payoff: speed. Computing energies, gradients, and NACs at multireference quality for thousands of trajectories is a clock-melting affair. With the learned NACs—and compatible ML models for energies/gradients—the workflow becomes hundreds of times faster. That lets you run bigger trajectory ensembles, shrink error bars, and finally test subtle mechanistic hypotheses that were previously buried in stochastic noise.
Are there caveats? Always. The method still relies on good training data that cover the relevant regions, especially around conical intersections. If your system flies apart or explores weird corners of configuration space, you’ll need careful active learning to keep the model honest. But the principle is clear: the right descriptor turns challenging ML problems into tractable ones.
Zooming out, this result nudges photodynamics toward a practical sweet spot: keep the trusted FSSH machinery, swap in machine learning where it’s physically justified, and spend the saved CPU time on real science.
MB
Reference
[1] J. Martinka, L. Zhang, Y.-F. Hou, M. Martyka, J. Pittner, M. Barbatti, P. O. Dral, A Descriptor Is All You Need: Accurate Machine Learning of Nonadiabatic Coupling Vectors, J. Phys. Chem. Lett. 16, 11732 (2025). 10.1021/acs.jpclett.5c02810