A bridge between molecular and solid-state photochemistry, enabling excited-state dynamics in extended systems.
In brief:
-
Introduces a NEWTON-X / CP2K interface for nonadiabatic dynamics with periodic boundary conditions.
-
Combines hybrid and semiempirical TDDFT methods to reach crystalline scales.
-
Demonstrates performance and accuracy on pyrazine molecules and crystals.
Modeling how light energy travels and dissipates in solids, nanostructures, and molecular crystals is one of the great frontiers of theoretical photochemistry. Until now, most nonadiabatic dynamics codes could describe only isolated molecules. Our new work—led by Anna Hehn from Christian-Albrechts-University Kiel,—changes that. By linking NEWTON-X, the widely used surface-hopping platform, with CP2K, the workhorse for density-functional simulations under periodic boundary conditions, they make photodynamics simulations of extended materials finally practical.
Surface hopping treats the nuclei as classical particles that occasionally jump between electronic states, mimicking how electrons and nuclei share energy after photoexcitation. This approach has been immensely successful for isolated molecules, but implementing it for solids requires efficient ways to compute excited states and their couplings across periodic boundaries.
The new interface offers exactly that. It supports several levels of electronic structure—from hybrid and generalized-gradient DFT to semiempirical schemes like sTDA and GFN1-xTB—and multiple forms of nonadiabatic couplings, including numerical orbital-derivative and phenomenological time-dependent Baeck–An terms. The result is a modular and flexible framework that can simulate hundreds of excited states at modest computational cost.
We validated the method on gas-phase pyrazine, a benchmark molecule for ultrafast internal conversion, reproducing known absorption spectra and relaxation lifetimes. Extending the same protocol to crystalline pyrazine, we computed broad electronic bands and tracked excited-state relaxation over 80 coupled states. Remarkably, a full trajectory step for a 40-atom unit cell requires only a few minutes on 64 cores, demonstrating that nonadiabatic dynamics in the solid state is no longer a distant dream.
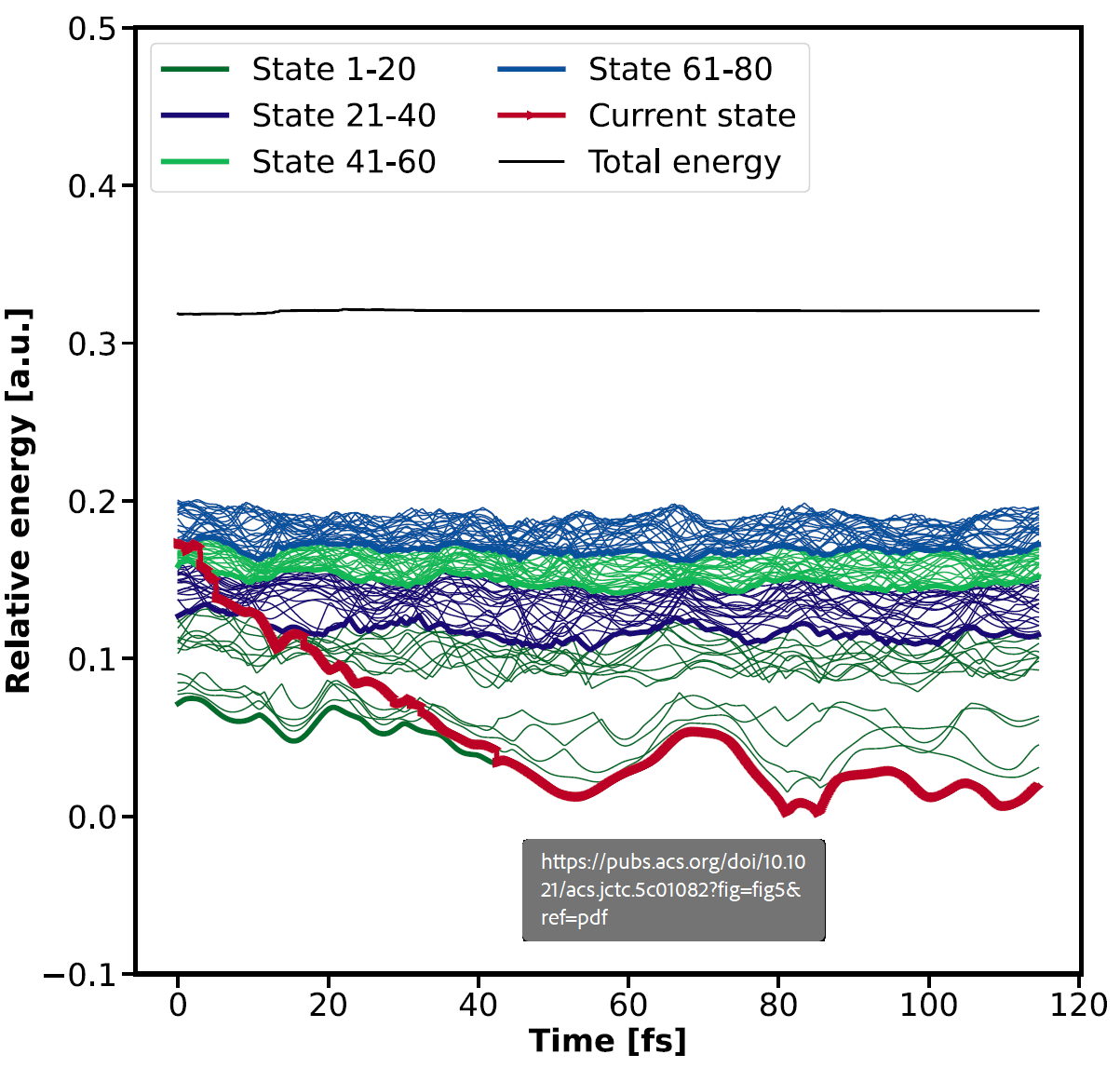
Exemplary trajectory for crystalline pyrazine based on orbital derivative couplings, an sTDA kernel, the PBE functional, and a MOLOPT-DZVP basis set. Excited states of higher energy are so dense that single trajectories cannot be distinguished. To nevertheless associate bands with a range of excited states, color coding is adjusted, always
highlighting 20 excited states with the same color.
Beyond pyrazine, this tool opens doors for studying charge transport in molecular semiconductors, exciton diffusion in light-harvesting networks, or the fate of photoexcited carriers in perovskites and organic LEDs. It’s a new playground where molecular photochemistry meets materials science.
MB
Reference
[1] J.-R. Vogt, M. Schulz, R. Souza Mattos, M. Barbatti, M. Persico, G. Granucci, J. Hutter, A. Hehn, A Density Functional Theory and Semiempirical Framework for Trajectory Surface Hopping on Extended Systems, J. Chem. Theory Comput. (2025). 10.1021/acs.jctc.5c01082