Simulations reveal a faster transfer step than previously thought.
In brief:
-
NM7HQ+, a strong photoacid, undergoes excited-state proton transfer (ESPT) to DMSO.
-
Ab initio molecular dynamics show the proton transfer occurs in <1 ps, not 2–3 ps as suggested by experiment.
-
The Eigen complex formed after transfer evolves slowly through rotational motion on the picosecond scale.
Proton transfer is a central process in chemistry and biology, occurring in enzymes, membranes, and light-sensitive chromophores. A special class of molecules, known as photoacids, becomes much stronger acids when electronically excited, allowing researchers to investigate proton motion on ultrafast timescales.
One such compound is N-methyl-7-hydroxyquinolinium (NM7HQ+}), a so-called super-photoacid. In its excited state, it is strong enough to transfer a proton even in aprotic solvents like dimethyl sulfoxide (DMSO), which lack the extended hydrogen-bond networks of water. This system provides a simple yet informative model for intermolecular excited-state proton transfer (ESPT).
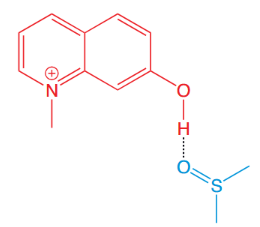
NM7HQ+-DMSO complex
Previous experiments indicated that ESPT from NM7HQ+ to DMSO occurred over 2–3 picoseconds. However, new ab initio molecular dynamics (AIMD) simulations challenge this interpretation.
In a project led by Niklas Sülzner, we used time-dependent density functional theory with a continuum solvent model to propagate 400 excited-state trajectories for 3 ps. The simulations show that the proton transfer itself is complete within less than 1 ps, forming an intermediate known as the Eigen complex.
The analysis of the trajectories highlights two stages. The first is the rapid proton transfer, occurring almost immediately after photoexcitation. The second is a slower evolution of the Eigen complex, not through diffusion, but via internal rotational motion of the DMSO and photoacid units. This separation process unfolds on the 1–3 ps timescale, explaining why earlier experiments detected a slower component and attributed it to the transfer itself.
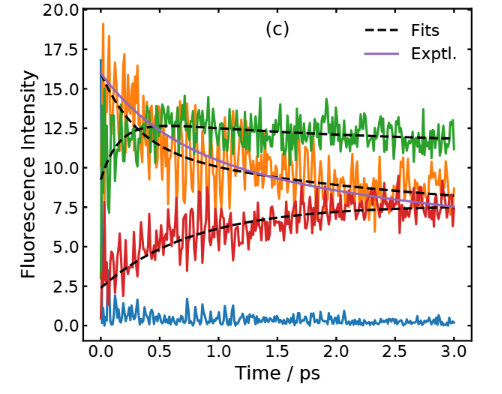
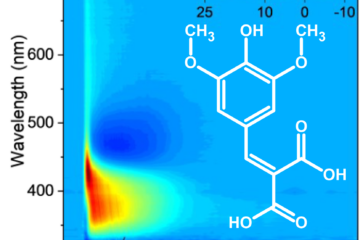
Theoretical reconstruction of the fluorescence intensity.
Comparison with fluorescence data supports this reinterpretation. The simulated time-dependent fluorescence decays reproduce the multi-exponential behavior observed experimentally. The fastest component (around 0.3 ps in the simulation) matches the rise of the Eigen complex, while the intermediate component (2 ps) corresponds to its slow reorganization. The agreement strengthens the conclusion that proton transfer is ultrafast, and the slower kinetics originate from structural relaxation.
This finding places NM7HQ+ near the boundary where ESPT is limited not by the intrinsic acidity of the donor, but by the solvent’s ability to reorganize. The short timescale observed (<1 ps) is comparable to solvation times in DMSO, suggesting that solvent dynamics already constrain the rate.
The work also highlights limitations. Using a polarizable continuum model avoids the complexity of explicit solvent molecules but misses specific interactions that could further modulate the dynamics. Extending the study to mixed quantum–classical (QM/MM) simulations would provide a more realistic representation of the local solvent environment.
Overall, these results refine our understanding of ESPT in non-aqueous media. Rather than a slow 2–3 ps transfer, the decisive step occurs in less than 1 ps, with subsequent dynamics dominated by the relaxation of the protonated complex. This provides a clearer mechanistic picture of how strong photoacids function in aprotic solvents, helping to interpret ultrafast spectroscopic data more accurately.
MB
Reference
[1] N. Sülzner, R. S. Mattos, M. Barbatti, Molecular Dynamics of the Ultrafast Excited-State Proton Transfer of the Super-Photoacid NM7HQ+ to the Aprotic Solvent DMSO, J. Phys. Chem. Lett. 16, 9124 (2025). 10.1021/acs.jpclett.5c02202