A community perspective on benchmarking and challenges of simulations beyond the Born-Oppenheimer approximation.
In brief:
-
This paper is a collective reflexion about the status of our nonadiabatic dynamics field.
-
It identifies four key photophysical phenomena as benchmark targets for nonadiabatic dynamics methods.
-
It surveys the full diversity of NAMD approaches—from exact quantum dynamics to AI-driven and hybrid techniques.
-
It launches a community-driven roadmap to define standards, share data, and unify practices.
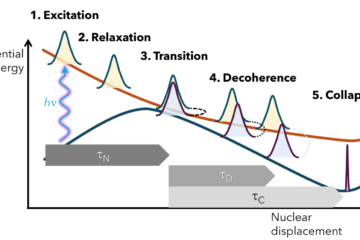
How do we know a simulation of excited-state dynamics is trustworthy?
That deceptively simple question is at the heart of a new perspective just published in J. Phys. Chem. A, co-authored by over 40 scientists from across the globe, including myself. The paper grew out of a CECAM workshop in Paris in 2024 and aims to bring clarity and structure to a field that’s as diverse as it is dynamic: nonadiabatic molecular dynamics.
This field studies what happens when light hits molecules and electrons and nuclei, jump out of equilibrium away from the Born-Oppenheimer approximation. The problem? We have dozens of methods to describe that phrnomenon—surface hopping, Ehrenfest, AIMS, MCTDH, CTMQC, RPMD, mapping formalisms, to name a few—and almost no standard way to compare them.
So we set out a roadmap.
At its core are four well-characterized types of light-induced molecular events:
-
Photoisomerization (e.g., retinal switching in vision),
-
Photodissociation (like NaI breaking under UV light),
-
Nonradiative relaxation (via conical intersections or intersystem crossing),
-
Excited-state proton transfer (as in fluorescent sensors).
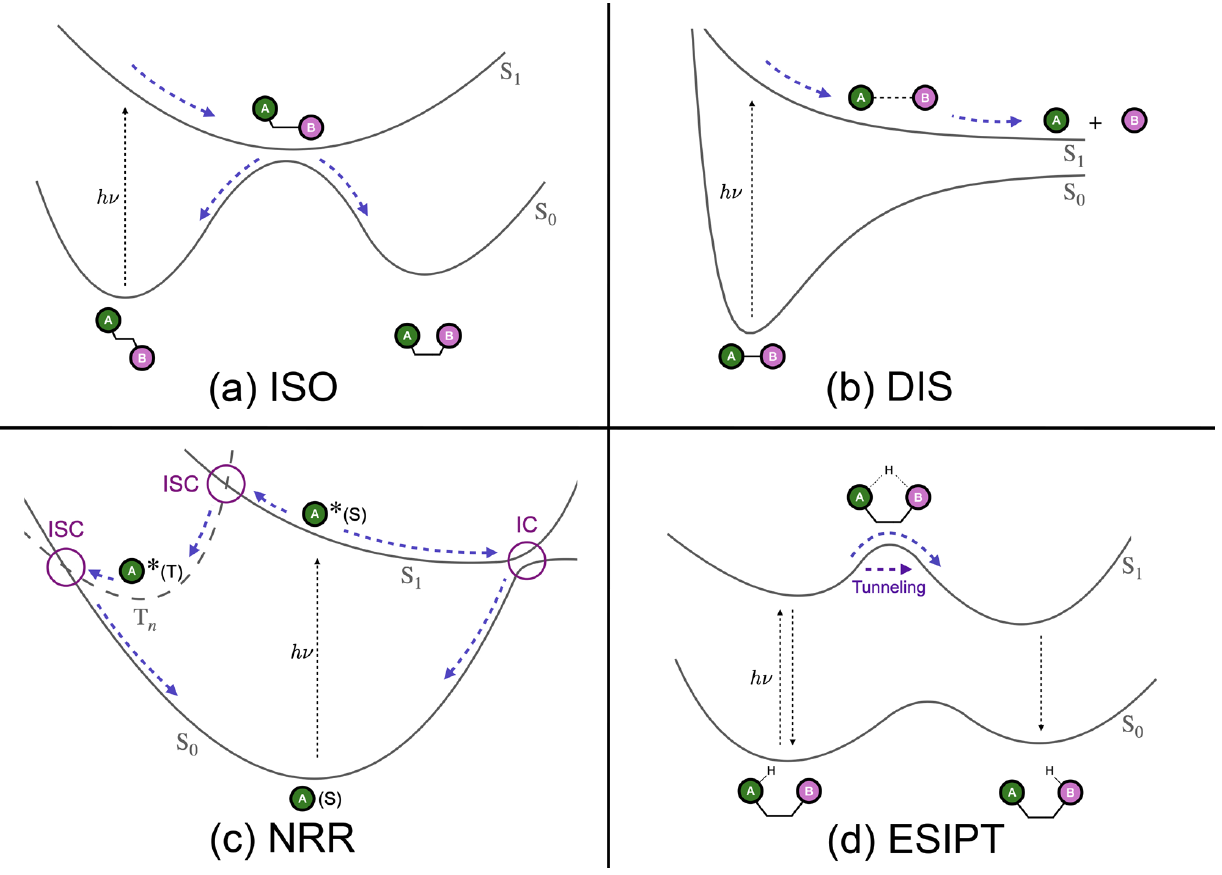
These cover most often cases in molecular photochemistry and photophysics, and offer fertile ground to compare methods, implementations, and approximations.
But benchmarks are tricky. What’s the right observable: populations? quantum yields? spectra? Should we compare to experiment or to nearly-exact simulations? And how do we ensure consistency when some methods need diabatic states, others adiabatic ones, and some avoid states altogether?
The paper doesn’t claim to solve all these issues, but it organizes them. It proposes a step-by-step community strategy, including:
-
agreed initial conditions,
-
standard test systems,
-
shared reference data,
-
and collaborative repositories for results and analysis tools.
And perhaps most importantly, it issues a call to work together across subfields. Whether you’re pushing tensor networks for wavepacket dynamics, or tuning spin-flip TDDFT for trajectory-based simulations, we need to talk the same language and test our tools side by side.
This roadmap isn’t just for method developers. It’s also for users, newcomers, and curious spectroscopists. Because only by building together can we turn today’s patchwork into tomorrow’s predictive theory.
MB
Reference
[1] L. L. E. Cigrang, B. F. E. Curchod, R. A. Ingle, A. Kelly, J. R. Mannouch, D. Accomasso, A. Alijah, M. Barbatti, W. Chebbi, N. Došlić, E. C. Eklund, S. Fernandez-Alberti, A. Freibert, L. González, G. Granucci, F. J. Hernández, J. Hernández-Rodríguez, A. Jain, J. Janoš, I. Kassal, A. Kirrander, Z. Lan, H. R. Larsson, D. Lauvergnat, B. Le Dé, Y. Lee, N. T. Maitra, S. K. Min, D. Peláez, D. Picconi, Z. Qiu, U. Raucci, P. Robertson, E. Sangiogo Gil, M. Sapunar, P. Schürger, P. Sinnott, S. Tretiak, A. Tikku, P. Vindel-Zandbergen, G. A. Worth, F. Agostini, S. Gómez, L. M. Ibele, A. Prlj, Roadmap for Molecular Benchmarks in Nonadiabatic Dynamics, J. Phys. Chem. A (2025). 10.1021/acs.jpca.5c02171