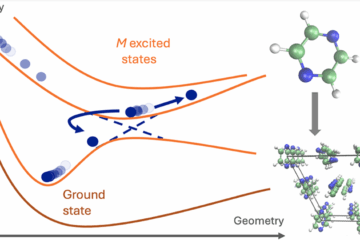
From radicals to Rydberg states: COLUMBUS is a veteran code that still does what others can’t.
In brief:
-
COLUMBUS offers unmatched flexibility for building custom multireference wavefunctions
-
Handles a wide spectrum of chemical challenges: radicals, Rydberg states, resonances, spin–orbit effects, and more
-
Compatible with Newton-X for nonadiabatic dynamics—an interface that’s been enabling simulations for two decades
If you’ve ever tried to model an excited molecule and ended up staring at an error message, you’re not alone. Molecular quantum chemistry is full of edge cases: diradicals, Rydberg states, ionic transitions, heavy elements with spin-orbit coupling, or bond dissociation. Most quantum chemistry codes handle these situations like picky eaters: they’ll work, but only if you feed them something very specific.
COLUMBUS is different.
It’s a multireference quantum chemistry program that doesn’t force your problem to fit its mold. Instead, it lets you reshape the wavefunction itself. Want to combine a valence active space with a few Rydberg orbitals? No problem. Need to simulate dissociation and still capture electron correlation? Done. Modeling bond formation on one end while another bond breaks elsewhere? Go ahead, COLUMBUS gives you the tools.
This flexibility shows up across the board in our recent collaborative paper surveying the COLUMBUS’ capabilities and illustrating its use for many different cases:
-
We tailored multiconfigurational wavefunctions for polyradicals and singlet/triplet crossings.
-
We simulated the curious fluorescence of hydrocarbon ion pairs.
-
We analyzed the elusive quintuple bond in uranium dimers.
-
We implemented complex absorbing potentials for electronic resonances.
-
And we applied spin-orbit configuration interaction to realistic photophysical systems.
Each case demanded something different and COLUMBUS adapted, thanks to its modular design and GUGA-based architecture.
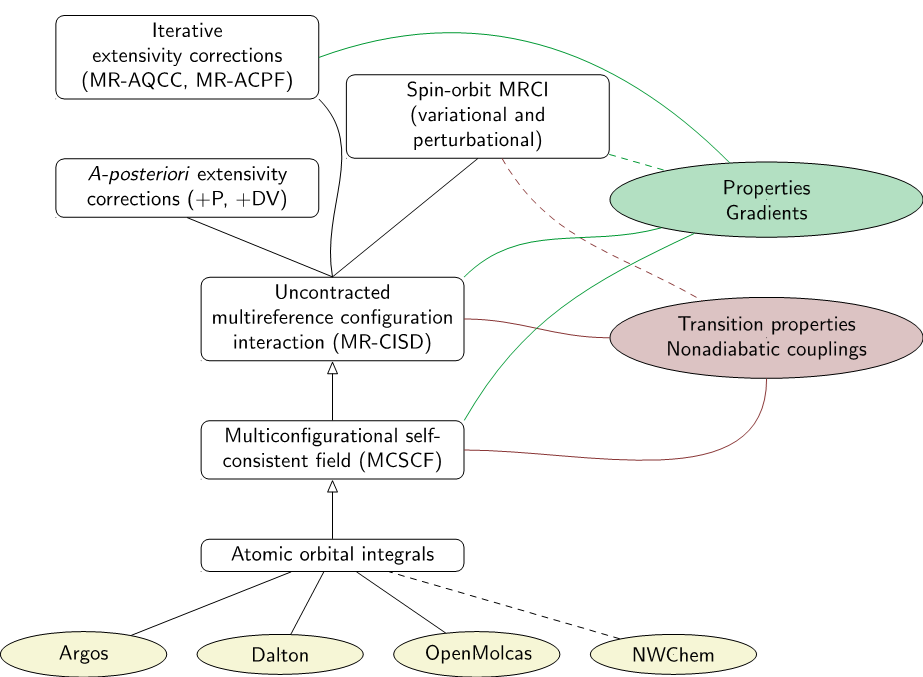
Summary of the functionality provided within COLUMBUS. Features in the release version are marked with solid lines, while features under development are marked with dashed lines.
Importantly, this isn’t limited to static calculations. COLUMBUS has had an interface to the Newton-X dynamics engine for nearly 20 years, making it one of the first high-level codes to support nonadiabatic molecular dynamics.
As a historical sidenote: in 2004, I was a postdoc with Hans Lischka, one of COLUMBUS’s main developers, working on MRCI simulations. Hans and I wanted to move toward nonadiabatic dynamics, which motivated me to start Newton-X. The interface to COLUMBUS, published in 2007, was the very first one I wrote.
Today, this interface, greatly optimized in Newton-X NS, continues to shine in complex scenarios. In Section 6.1 of the paper, Saikat Mukherjee and I showed how to use it with customized active spaces that capture complex photodynamics at manageable cost.
We applied this strategy to excited ethylene (including Rydberg states and bond-breaking) and cyclobutanone, where dissociation pathways emerge from a Rydberg-excited state. And we’ve gone a step further: using COLUMBUS as a backend to generate machine learning training data. In one case, 6000 CASSCF points trained a model that can now simulate photodynamics at quantum accuracy, but thousands of times faster.
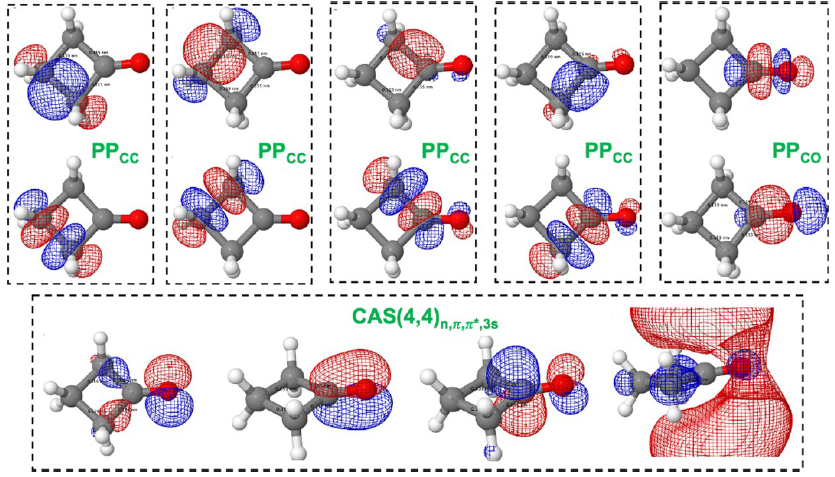
Example of generalized orbital active space able to describe arbitrary dissociations in cyclobutanone.
The take-home message? COLUMBUS doesn’t just solve equations, it adapts to your chemistry. Whatever weird, beautiful, or stubborn system you’re working on, chances are COLUMBUS can handle it. And thanks to decades of steady development, it’s ready to keep doing so.
MB
Reference
[1] F. Plasser, H. Lischka, R. Shepard, P. G. Szalay, R. M. Pitzer, R. L. R. Alves, A. J. A. Aquino, J. Autschbach, M. Barbatti, J. R. Carvalho, J. C. V. Chagas, L. González, A. Hansen, B. Jayee, M. Kertesz, F. B. C. Machado, S. Matsika, S. A. do Monte, S. Mukherjee, D. Nachtigallová, R. Nieman, V. P. Oliveira, M. Oppel, C. A. Parish, J. Pittner, L. G. F. dos Santos, A. Scrinzi, M. K. Sit, R. F. K. Spada, M. Thodika, D. C. A. Valente, Á. Vázquez-Mayagoitia, E. Ventura, J. Westermayr, A. Zaichenko, Z. Zhang, COLUMBUS─An Efficient and General Program Package for Ground and Excited State Computations Including Spin–Orbit Couplings and Dynamics, J. Phys. Chem. A (2025). 10.1021/acs.jpca.5c02047