A new AI-powered protocol learns excited states with accuracy that finally makes it practical.
In brief:
-
A novel multi-state neural network (MS-ANI) learns any number of electronic states across different molecules.
-
Gap-driven dynamics (gapMD) ensure critical regions like conical intersections are well-sampled.
-
Active learning potentials based on the combination of these methods yielded accurate dynamics for several different molecules.
Simulating what molecules do after absorbing light is a cornerstone of photophysics and photochemistry. But doing this accurately has always been painfully expensive. You need to compute excited-state properties for every nuclear configuration along a trajectory, hundreds of thousands of times! Machine learning (ML) has been a promising way to speed up this process, by replacing expensive electronic structure evaluations. Nevertheless, training accurate ML models for nonadiabatic dynamics has been an enormous challenge.
In our latest paper, led by Martyka and Dral, we introduce a robust and fully automated ML-based protocol that learns to simulate excited-state dynamics across different molecules. It delivers the speed we need, the accuracy we want, and none of the old instability headaches.
The novelty is a new kind of ML model: the multi-state ANI neural network (MS-ANI). Unlike earlier approaches that trained separate models for each electronic state or lumped everything into one big output layer, our model takes in both the molecular geometry and the state label (like “first excited state”). That lets it learn inter-state relationships—like energy gaps—right from the start.
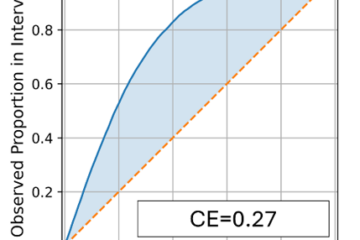
In tests on molecules like pyrene, the model predicted excited-state energies and gaps with chemical accuracy—better than traditional single- or multi-output networks (see Fig. 1 from the paper).
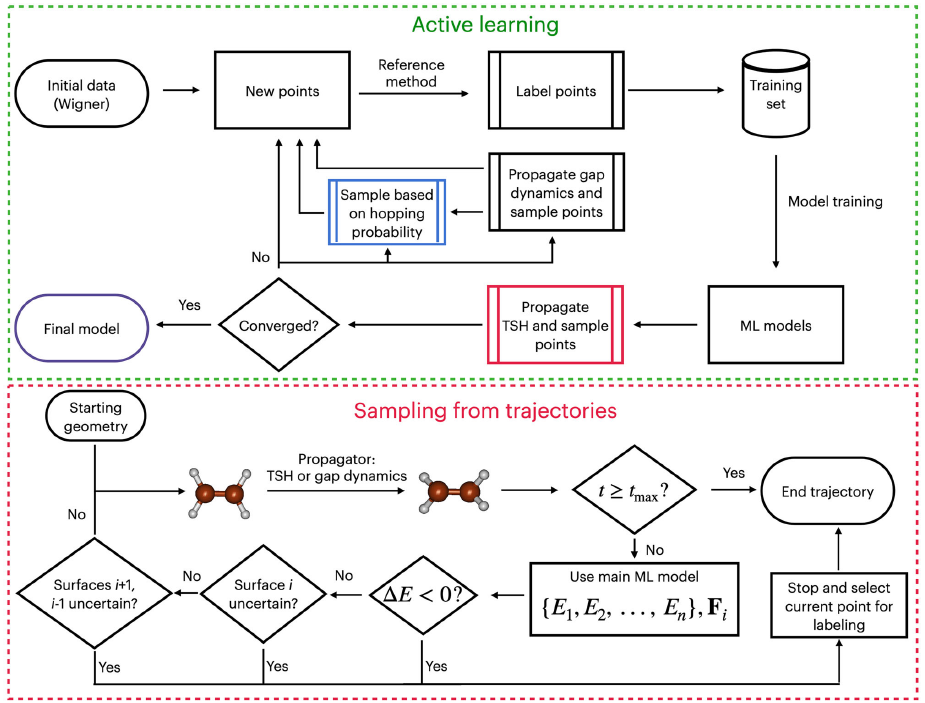
Flowchart of active learning with MS-ANI and gapMD algorithms,
But even the best model needs good training data. And the tricky parts of potential energy surfaces—like conical intersections—are usually underrepresented.
To fix this, we introduced gap-driven molecular dynamics (gapMD). It’s a clever way to steer simulations directly into regions with small energy gaps, ensuring these hotspots are well-sampled and learned.
All of this is embedded in a full active learning loop. It selects training points automatically, tunes uncertainty thresholds statistically (no fiddling with knobs), and keeps improving until the model gets things right.
We tested the full pipeline on several cases:
-
Fulvene, where we matched reference quantum dynamics with 10x fewer points and 30x less time.
-
Ferro-wire, a big 80-atom system with multiple states, trained in only three iterations.
-
Azobenzene, the classic photoswitch, where long trajectories revealed cis-trans oscillations after internal conversion that had been missed before.
Active learning based on MS-AMI with gapMD is definitely a huge step forward in establishing robust ML protocols for excited-state dynamics.
MB
Reference
[1] M. Martyka, L. Zhang, F. Ge, Y.-F. Hou, J. Jankowska, M. Barbatti, P. O. Dral, Charting electronic-state manifolds across molecules with multi-state learning and gap-driven dynamics via efficient and robust active learning, npj Comput. Mater. 11, 132 (2025). 10.1038/s41524-025-01636-z