Effortlessly achieve rotationally invariant machine learning of vectors with the rotate-predict-rotate (RPR) method.
In brief:
- This work introduces the rotate-predict-rotate (RPR) method, ensuring rotational covariance for machine learning of vector and tensor properties.
- RPR achieves high accuracy and efficiency by aligning molecules to a canonical orientation based on their inertia tensor, predicting properties, and then rotating back.
- It was successfully tested on 1,2-dichloroethane, significantly improving dipole moment and polarizability predictions with a small training set.
- RPR provides a computationally efficient alternative to complex equivariant machine learning architectures and is suitable for large-scale molecular dynamics and spectroscopy.
A Simple Solution for Machine Learning of Vector Quantities
We are excited to share that our latest paper, A Simple Approach to Rotationally Invariant Machine Learning of a Vector Quantity, has been accepted in The Journal of Chemical Physics. This work, a collaboration led by Jiri Pittner‘s group, tackles a long-standing challenge in molecular simulations: how to efficiently and accurately predict vector properties, like dipole moments, using machine learning (ML) while ensuring they remain consistent under rotations.
Why Is This Important?
Predicting scalar properties like potential energy with ML is relatively straightforward because scalars are invariant under rotations. However, things get tricky with vector (and tensor) properties, which change their components depending on the molecule’s orientation. Traditional ML methods struggle with this, often requiring complex adjustments to guarantee the correct behavior under rotations.
Many existing techniques embed rotational equivariance directly into the machine learning architecture, but these approaches can be computationally expensive. This is where our work steps in, offering a simpler, faster solution without sacrificing accuracy.
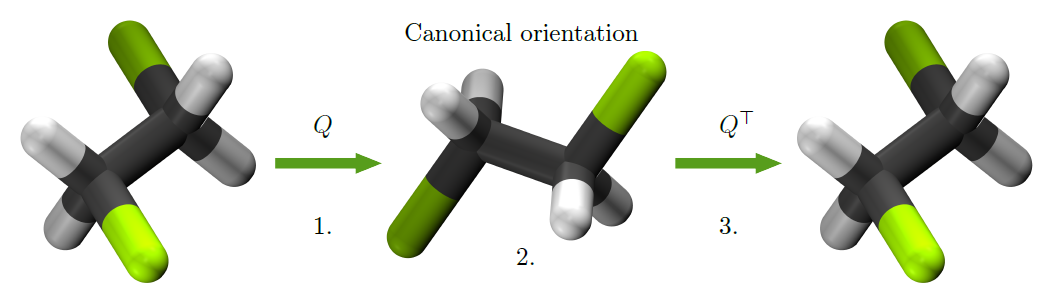
The Rotate-Predict-Rotate (RPR) Method
Our approach is called rotate-predict-rotate (RPR). The idea is simple: instead of redesigning the ML architecture, we focus on manipulating the molecular system itself. Here’s how it works:
- Rotate: First, we align the molecule to a “canonical” orientation, defined by the principal axes of the molecular inertia tensor.
- Predict: We train and predict vector properties (like dipole moments) in this standardized orientation.
- Rotate Back: Finally, we transform the predicted vectors back into the molecule’s original orientation.
This process ensures that the predicted vectors correctly transform under rotations, keeping the symmetry intact.
Why This Matters
The beauty of RPR is its simplicity and efficiency. Instead of requiring complex model designs, the RPR method only involves elementary linear algebra (matrix rotations), making it computationally light. It also speeds up training and prediction, allowing us to handle larger datasets efficiently—a critical advantage when using active learning techniques.
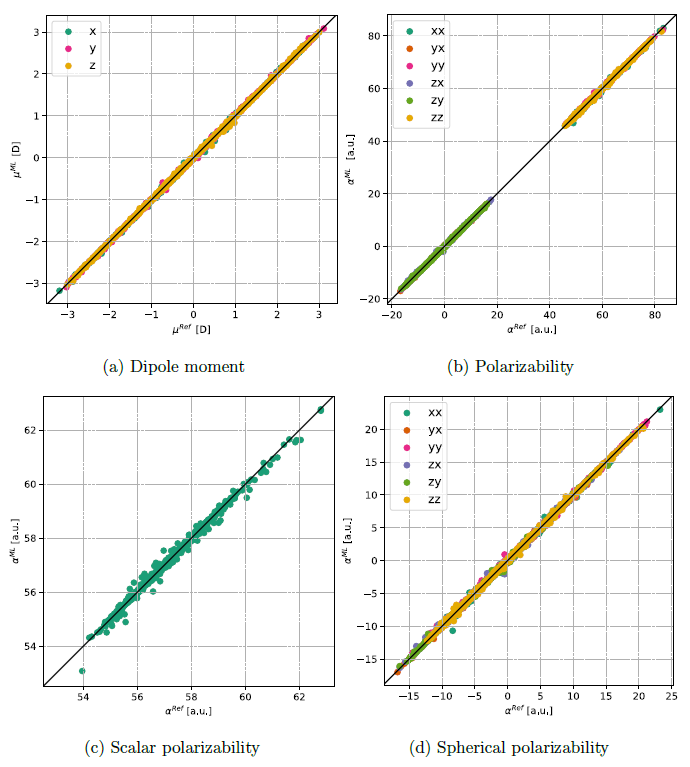
We tested this method on 1,2-dichloroethane, a system where the dipole moment is highly sensitive to molecular orientation. By enhancing molecular descriptors with chlorine atom coordinates, we significantly improved the accuracy of our predictions. Even with a modest training set of 1000 points, the RPR method produced excellent results for both dipole moments and polarizabilities.
Fast, Accurate, and Flexible
The RPR method is not only accurate but also highly scalable. It can be easily extended to tensor properties like polarizabilities and can handle large molecular systems. We showed that the method achieves impressive computational speeds, even on single-core processors, while maintaining high accuracy.
Our approach stands out because it offers a straightforward way to ensure rotational covariance in machine learning models without the overhead of more intricate ML architectures. Whether you’re working on molecular dynamics, spectroscopy, or material design, RPR provides a practical solution for predicting vector and tensor properties.
What’s Next?
This work opens up exciting new possibilities for machine learning in computational chemistry. With its simplicity and efficiency, the RPR method is a powerful tool that can be applied to a wide range of problems. We’re excited to see how this technique will be used and refined in the future.
MB
Reference
[1] J. Martinka, M. Pederzoli, M. Barbatti, P. O. Dral, J. Pittner, A simple approach to rotationally invariant machine learning of a vector quantity, J. Chem. Phys. 161, 174104 (2024). DOI: 10.1063/5.0230176