QDCT seamlessly enhances surface hopping accuracy without adding computational cost.
In brief:
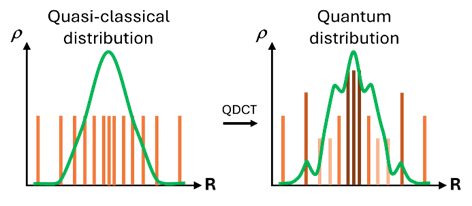
- Innovative Method: Quantum Dynamics from Classical Trajectories (QDCT) recovers quantum wavepacket behavior from classical trajectories generated by surface hopping (TSH), improving simulation accuracy.
- No Extra Cost: The method upgrades TSH results to the ab initio multiple spawning (AIMS) level without requiring additional computational resources or new electronic calculations.
- Error Correction: QDCT corrects coherence errors in TSH, leading to more reliable and accurate molecular dynamics simulations. QDCT also diagnostics potential failures in TSH.
- Versatility: Applicable across various surface hopping methods, both stochastic and deterministic, making QDCT widely useful.
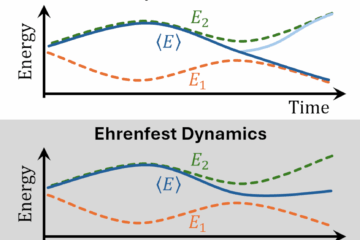
Nonadiabatic molecular dynamics, where electronic and nuclear motions are intricately coupled, is essential for understanding the time evolution of molecular systems. Among the computational methods designed to do such simulations, Trajectory Surface Hopping (TSH) stands out. TSH effectively balances computational efficiency with accuracy by treating the nuclear motion as a swarm of independent classical trajectories while describing nonadiabatic events by allowing each trajectory to switch between electronic states occasionally. However, TSH has its limitations, particularly in neglecting nonlocal nuclear quantum effects, which can lead to inaccuracies in the simulations.
In a significant advancement, Rafael S. Mattos, in collaboration with Saikat Mukherjee and me, introduced a novel methodological protocol called Quantum Dynamics from Classical Trajectories (QDCT). This method, which is a core component of Rafael’s PhD thesis, checks the quality and enhances the accuracy of TSH by reconstructing quantum wavepacket behavior from the classical trajectories generated by TSH. Notably, QDCT achieves this without requiring additional computational costs, making it a highly efficient and practical tool for researchers.
Trajectory Surface Hopping: The Current State
TSH is widely used in molecular dynamics because it allows for the incorporation of nonadiabatic effects through (usually stochastic) hops between potential energy surfaces. While effective, TSH’s classical treatment of nuclei means that it cannot fully capture nuclear quantum effects such as tunneling or zero-point energy. These limitations can lead to errors in specific simulations, particularly in cases where nonlocal quantum effects play a significant role.
The Innovation of QDCT
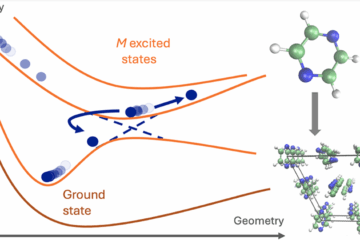
QDCT represents a breakthrough that allows researchers to recover quantum wavepacket information from the classical trajectories produced by TSH. Essentially, QDCT acts as a post-processing step, refining the results of TSH to achieve accuracy comparable to more sophisticated quantum methods like the Ab Initio Multiple Spawning (AIMS) approach.
QDCT integrates AIMS equations of motion using TSH points as a grid and creating virtual trajectories to allow spawning and interpolation between TSH trajectories. The virtual-trajectories algorithm is the heart of QDCT and constitutes one of its main innovations.
One of QDCT’s most valuable features is its ability to identify and correct potential issues in TSH simulations. For example, TSH can sometimes overestimate quantum coherence, leading to inaccurate results. QDCT can correct for this over-coherence, thus improving the reliability of the simulation. This correction enhances both the accuracy and the confidence researchers can have in their results.
Efficiency and Versatility
The most compelling aspect of QDCT is its efficiency. Since the method does not require new electronic structure calculations, it adds virtually no computational cost to the original TSH simulation. This makes QDCT an attractive option for researchers looking to improve their simulations without a significant increase in computational overhead.
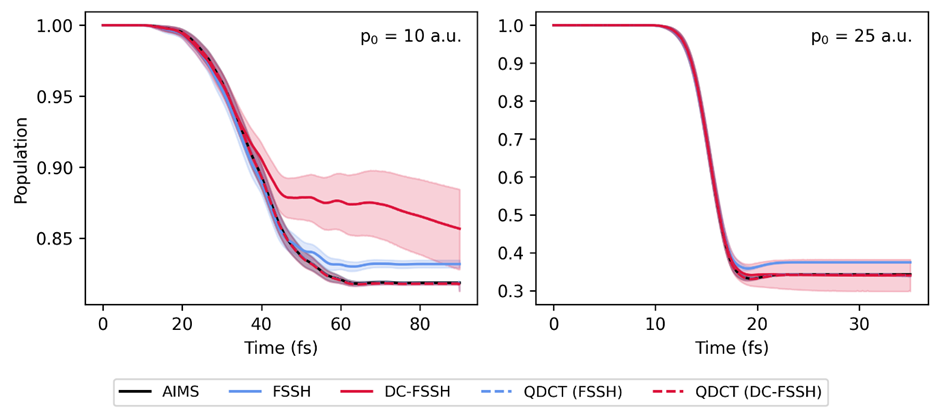
Moreover, QDCT is not limited to the fewest switches surface hopping (FSSH); it can be applied to various other surface hopping methods, whether stochastic or deterministic. This versatility means that QDCT has the potential to become a widely used tool in the study of nonadiabatic processes across different types of molecular systems.
Looking Forward
Rafael’s work on QDCT is a significant contribution to the field of nonadiabatic molecular dynamics. By enabling the recovery of quantum wavepacket behavior from classical trajectories, QDCT offers a robust and efficient way to verify and enhance the accuracy of TSH simulations. As the method continues to be refined and applied to more complex systems, it promises to provide deeper insights into the quantum dynamics that drive nonadiabatic molecular processes.
MB
Reference
[1] R. Souza Mattos, S. Mukherjee, M. Barbatti, Quantum Dynamics from Classical Trajectories, J. Chem. Theory Comp. (2024). DOI: 10.1021/acs.jctc.4c00783