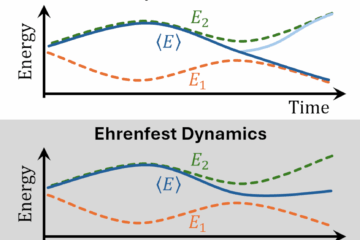
Mapping between probability distributions can save a lot of time in dynamics and spectrum simulations.
In brief:
- Importance sampling (IS) estimates properties for a particular distribution, using results calculated for another distribution.
- With IS, spectrum and dynamics obtained for a set of initial conditions are transformed into those for another set at negligible cost.
- IS was implemented in Newton-X and tested to transform results computed at a particular temperature to different target temperatures.
In dynamics and spectrum simulations, mean values for several observables are computed for an ensemble of points distributed in the nuclear phase space, each point corresponding to a different set of nuclear geometry and momentum. The mean value of an observable f computed at point Xl (which is distributed according to P) is
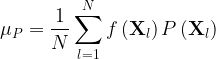
where N is the number of points.
Most of the computational cost is in the calculation of f, which is usually some electronic property like excitation energy or oscillator strength. To make it worse, such type of calculation must be repeated for all N points.
Suppose you’ve computed the mean value of f for phase space points distributed at a temperature of 300 K, and, now, you also need to know the mean value at 500 K. How would you proceed?
The trivial way is just to get Xl distributed at the new temperature and recalculate all N values of f again. Of course, the computational cost is doubled.
But there is another much cheaper way of getting this new mean value.
If the new distribution is Q, we can rewrite the mean values as
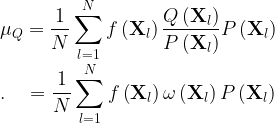
Therefore, if you know how to compute the weights ω = Q/P for each point, you can just use those f you had calculated before for P to get μQ.
And, in fact, in many situations, we know how to compute ω: this occurs when the points are sampled with an analytical distribution, like a Wigner function for instance.
Another way of looking at this procedure is that you can calculate the mean value of f for the distribution Q using the original distribution P if, instead of averaging f, you average g = fω:
![\displaystyle \mu_Q = \frac{1}{N}\sum_{l=1}^{N} \left[ f\left(\mathbf{X}_l \right) \omega\left(\mathbf{X}_l \right) \right] P\left(\mathbf{X}_l \right) \\ .\quad = \frac{1}{N}\sum_{l=1}^{N} g\left(\mathbf{X}_l \right) P\left(\mathbf{X}_l \right)](https://s0.wp.com/latex.php?latex=%5Cdisplaystyle+%5Cmu_Q+%3D%C2%A0%5Cfrac%7B1%7D%7BN%7D%5Csum_%7Bl%3D1%7D%5E%7BN%7D+%5Cleft%5B+f%5Cleft%28%5Cmathbf%7BX%7D_l+%5Cright%29+%5Comega%5Cleft%28%5Cmathbf%7BX%7D_l+%5Cright%29+%5Cright%5D+P%5Cleft%28%5Cmathbf%7BX%7D_l+%5Cright%29+%5C%5C+.%5Cquad+%3D%C2%A0%5Cfrac%7B1%7D%7BN%7D%5Csum_%7Bl%3D1%7D%5E%7BN%7D+g%5Cleft%28%5Cmathbf%7BX%7D_l+%5Cright%29+P%5Cleft%28%5Cmathbf%7BX%7D_l+%5Cright%29++&bg=ffffff&fg=000&s=1&c=20201002)
This technique of using a mapping between two distributions — called importance sampling — has been implemented and tested by Fabris Kossoski for dynamics and spectrum simulations with Newton-X [1].
The figure below illustrates the IS results for the absorption spectrum of phenol. In each panel, it is shown the spectrum calculated at a specific source temperature Ts, and its IS estimate for two target temperatures Tt.
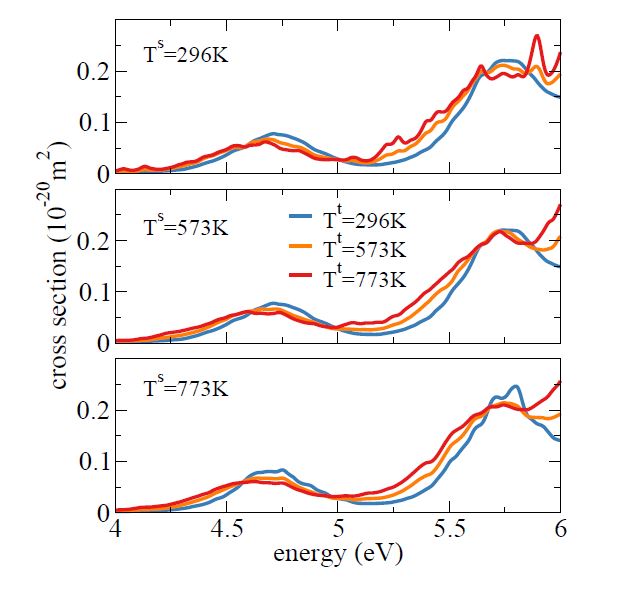
For instance, in the upper panel, the spectrum was calculated for initial conditions sampled with a Wigner distribution for Ts = 296 K (blue). Then, IS was used to transform this spectrum into the spectra at Tt = 573 K (orange) and Tt = 773 K (red). Just to emphasize, these two later spectra did not demand any new electronic structure calculations, only the calculation of the ω weights, whose cost is negligible.
Now, compare the spectra at 296 K (blue curves) in the three panels. The one in the upper panel is computed from first principles; in the other two, they were computed via IS. We can see that the overall agreement is pretty good. As expected, we can also see that the deviation from the first-principles result is larger for Ts = 773 (bottom) than for Ts = 573 K (middle).
Details on the implementation of IS and test results for dynamics and spectrum are discussed in Ref. [1]. The IS algorithm will be soon released in a new version of Newton-X.
MB
Reference
[1] F. Kossoski and M. Barbatti, Nuclear Ensemble Approach with Importance Sampling; J. Chem. Theory Comp., doi: 10.1021/acs.jctc.8b00059 (2018).