Topographical analysis of the dimer’s excited state shows that internal conversion after first proton transfer blocks the stepwise process.
This year, it completes the 20th anniversary of the seminal paper where Ahmed Zewail and his team, based on time-resolved spectroscopy, proposed that the photo-induced double proton transfer in 7-Azaindole (7AI) dimer is a stepwise process [1].
Since then, this paper has motived an uncommonly fierce debate on the nature of this transfer—whether it is really stepwise or alternatively concerted [2,3].
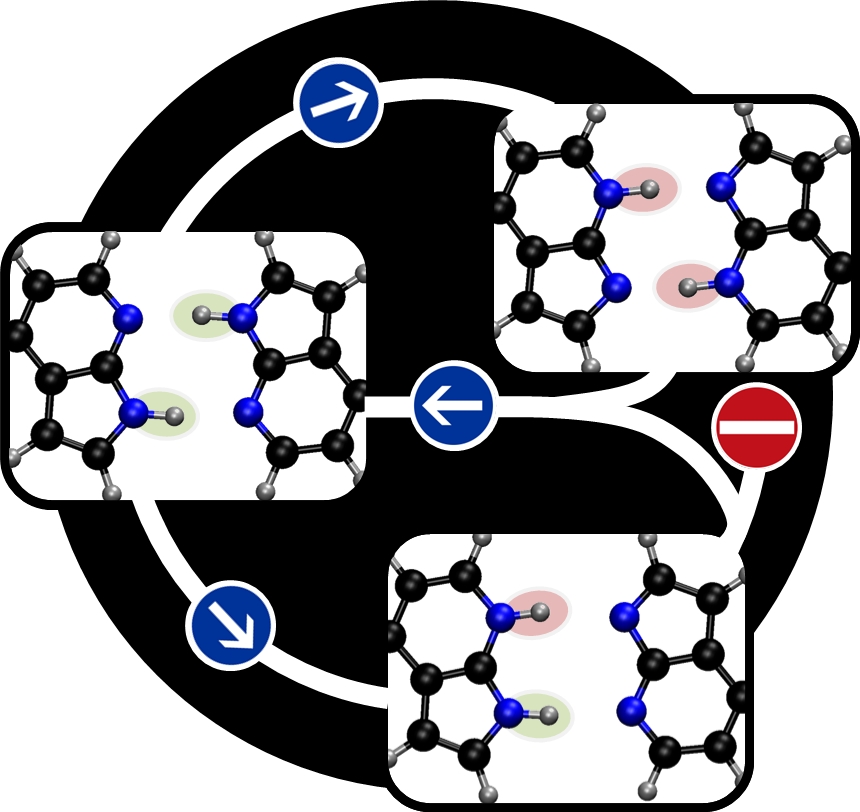
The excited state double proton transfer in 7AI dimer can in principle follow through two different mechanisms: either concerted (both protons jump to the other side about at the same time) or stepwise (one proton jumps first, then the other one does). We show that the stepwise mechanism is kinetically and energetically unfavorable.
Topographical analysis of the dimer’s excited state shows that internal conversion after first proton transfer blocks the stepwise process.
Using high-level computational simulations of static and dynamic properties based on CC2 and ADC(2) methods, Rachel Crespo-Otero (London), Nawee Kungwan (Chiang Mai), and I found out that much of these earlier discussions was induced by inappropriate theoretical modeling, which led to biased interpretation of the experimental results. These findings just appeared in a recent Edge Article of the Chemical Science [4].
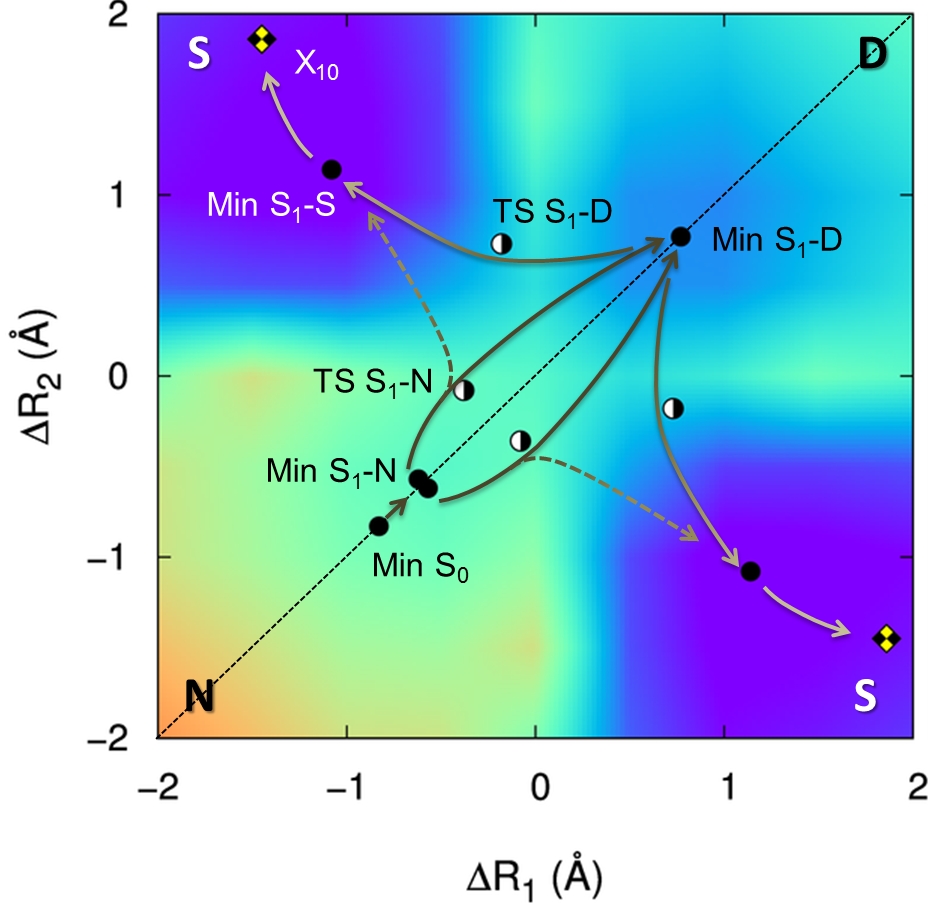
We show that earlier models (based on CIS, CASPT2, MRMP, and TDDFT) provided either a wrong or incomplete topographic description of the excited-state potential energy surface of 7AI dimer. They delivered a bad balance between the energies of the local-excitation and charge-transfer regions, and completely missed the possibility of internal conversion to the ground state.
A proper topographic description of the PES clearly reveals that the stepwise mechanism is not accessible due to kinetic and thermodynamic reasons.
Created with GIMP
Single proton transfer can occur (N to S in the figure above), but when it does, an energy barrier blocks the transfer of the second proton (S to D) and the dimer relaxes through internal conversion at the X10 conical intersection. Thus, the stepwise process is inhibited and the double proton transfer is left with a single possibility: to occur through a concerted mechanism (N to D).
This case-study illustrates how computational simulations may sometimes lead to unphysical interpretation of experimental results.
The movie below, result from surface hopping simulations, shows how excited 7AI dimer does the double PT in the ballistic regime, when tunneling is negligible.
References
[1] A. Douhal, S. K. Kim and A. H. Zewail, Femtosecond molecular dynamics of tautomerization in model base pairs, Nature 378, 260 (1995).
[2] J. Catalan, P. Perez, J. C. del Valle, J. L. G. de Paz and M. Kasha, H-bonded N-heterocyclic base-pair phototautomerizational potential barrier and mechanism: The 7-azaindole dimer, Proc. Natl. Acad. Sci. USA 101, 419 (2004).
[3] S. Takeuchi and T. Tahara, The answer to concerted versus step-wise controversy for the double proton transfer mechanism of 7-azaindole dimer in solution, Proc. Natl. Acad. Sci. USA 104, 5285 (2007).
[4] R. Crespo-Otero, N. Kungwan, M. Barbatti, Stepwise Double Excited-State Proton Transfer Is Not Possible in 7-Azaindole Dimer, Chem. Sci., doi:10.1039/C5SC01902H (2015)



0 Comments