

MD simulations can be 10x faster by replacing files with socket communication.
In brief:
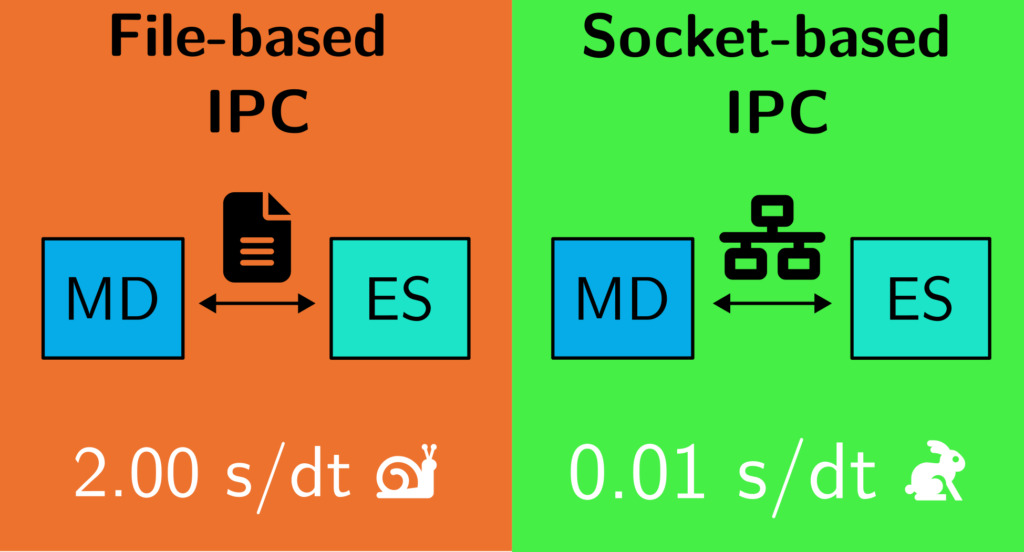
- Efficient Data Exchange: Socket-based IPC replaces traditional file-based communication, significantly reducing data transfer times in molecular dynamics simulations.
- Tenfold Speed Improvement: Achieved faster MD timesteps by minimizing disk I/O operations and streamlining energy and gradient evaluations with a client-server architecture.
- Broad Applicability: The approach supports more extensive simulations and reduces computational costs, paving the way for new advancements in nonadiabatic dynamics and machine learning integration.
Our latest publication in the Journal of Physical Chemistry Letters introduces a groundbreaking approach to enhancing molecular dynamics (MD) simulations through the use of socket-based inter-process communication (IPC). By replacing traditional file-based data exchanges with direct socket communications, we achieved dramatic speed improvements in simulations involving Newton-X interfaced with MLatom.
The fundamental challenge of MD simulations lies in their reliance on precise, iterative calculations of potential energy surfaces (PES) to predict the evolution of molecular systems. Traditionally, this involves extensive file-based data exchanges between MD and electronic structure (ES) programs, slowing down simulations due to frequent disk operations. As computational advances accelerate PES calculations—especially with the rise of machine learning potentials—the bottleneck of data exchange has become increasingly evident.
To address this, Matheus Bispo implemented a socket-based client-server architecture. In this model, Newton-X acts as a client, continuously requesting energy and gradient data, while MLatom serves as the ES server, computing and delivering results via network sockets.
This configuration minimizes the need for disk I/O operations and significantly reduces computational delays associated with frequent program startups. The results? A staggering tenfold reduction in computational time per timestep compared to the traditional file-based approach.
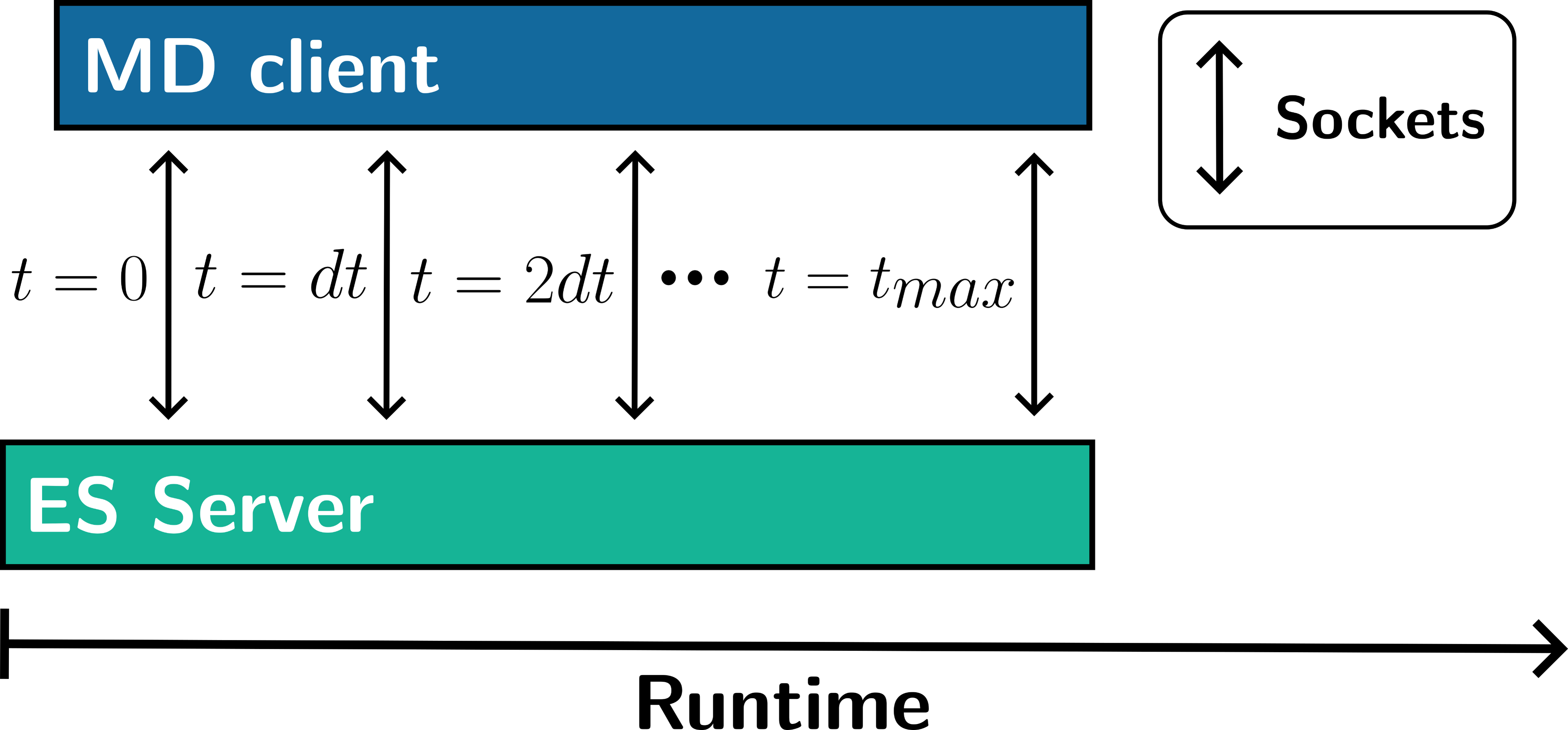
The implications of this improvement extend far beyond mere speed enhancements. With more efficient data handling, MD simulations can explore longer timescales and higher throughput scenarios. This opens new possibilities for studying complex molecular dynamics in real time, accelerating research across fields such as photophysics, chemical reactions, and material science.
Our proof-of-concept tests used surface hopping trajectories for fulvene—a standard molecule in nonadiabatic studies. We observed consistent improvements, with socket-based IPC reducing the computational time per timestep from over 2.6 seconds (file-based) to a mere 0.012 seconds. This leap in efficiency not only cuts down computational costs but also significantly lowers the carbon footprint of large-scale MD simulations, aligning with sustainability goals.
As computational chemistry moves towards integrating machine learning potentials with traditional quantum mechanical methods, our socket-based communication framework presents a scalable and versatile solution. We invite the MD and broader computational science community to explore this approach, which promises to revolutionize simulation workflows through enhanced speed, accuracy, and environmental consciousness.
MB
Reference
[1] M. de O. Bispo, M. Barbatti, Accelerating Molecular Dynamics Simulations Using Socket-Based Inter-Process Communication, J. Phys. Chem. Letters 15, 11891 (2024). 10.1021/acs.jpclett.4c02860