
Quantum mechanical and machine learning dynamics with MLatom
In brief:
- We introduce MLatom@XACS, an open-source software ecosystem for on-the-fly surface hopping nonadiabatic dynamics using the Landau–Zener–Belyaev–Lebedev (LZBL) algorithm.
- The software supports a wide range of quantum mechanical and machine learning methods, including the ready-to-use AIQM1.
- Example scripts provided streamline the workflow from geometry optimization to final result plotting, making advanced simulations accessible to a broader public.
In theoretical chemistry, the ability to quickly model and predict molecular behavior during electronic transitions is crucial. Pavlo Dral’s latest development, the MLatom@XACS software ecosystem, which we have contributed to, aims to advance this field significantly. This open-source platform is designed for on-the-fly surface hopping nonadiabatic dynamics, using the Landau–Zener–Belyaev–Lebedev (LZBL) algorithm.
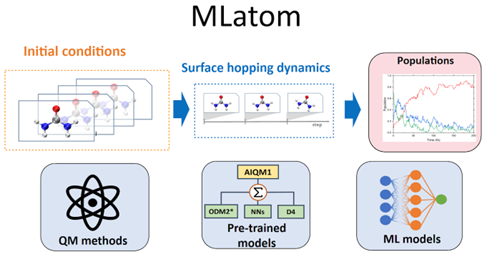
Versatility and Accessibility Through Python
MLatom@XACS stands out for its versatility and ease of use. It offers a Python API, allowing users to seamlessly integrate a wide range of quantum mechanical (QM) and machine learning (ML) methods into their simulations. Whether you’re using ab initio QM methods like CASSCF and ADC(2) or semi-empirical QM methods such as AM1, PM3, OMx, and ODMx, MLatom@XACS supports them all. Additionally, it is compatible with various ML potentials, including KREG, ANI, and MACE.
Customizable and Ready-to-Use Methods
One of the central features of MLatom@XACS is its flexibility in method combinations. Users can build their combinations of QM and ML methods tailored to their specific research needs. For those who prefer ready-to-use solutions, we provide AIQM1, an out-of-the-box method based on Δ-learning.
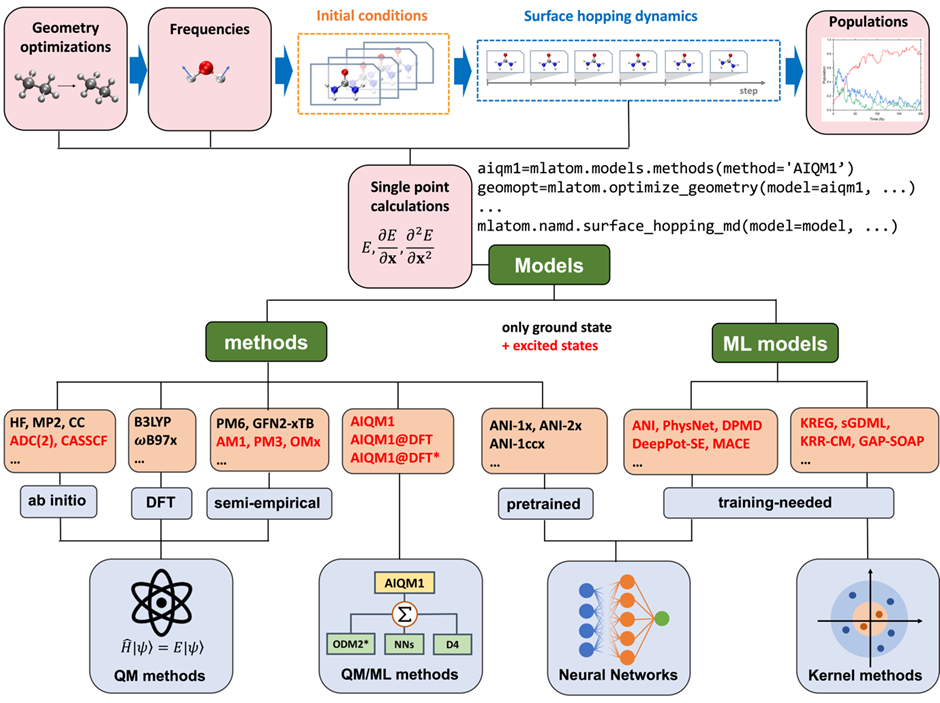
Simplified Workflow with Example Scripts
To further simplify the user experience, we have included example scripts that streamline the entire workflow. With just a few dozen lines of code, users can achieve comprehensive results from initial geometry input to final population plots. These scripts handle critical tasks such as geometry optimization, normal mode calculations, initial condition sampling, parallel trajectory propagation, population analysis, and final result plotting. This user-friendly approach makes sophisticated nonadiabatic dynamics simulations accessible to a broader public, including those who may not be experts in computational chemistry.
Integration and Future Enhancements
MLatom@XACS is not just a standalone tool; it is designed to be an integral part of broader ML model training protocols for nonadiabatic dynamics. Its compatibility with various ML models ensures that users can seamlessly incorporate it into their existing workflows. Looking ahead, we envision an even deeper integration with Newton-X. This integration aims to introduce a wide array of functionalities, such as fewest-switch surface hopping, thus enhancing the capabilities of MLatom@XACS and facilitating similar workflows via the Python API.
Conclusion
The MLatom@XACS software ecosystem represents a significant advancement in the field of nonadiabatic dynamics. Its combination of versatility, ease of use, and powerful capabilities makes it an invaluable tool for researchers. By providing both customizable and ready-to-use methods, along with comprehensive example scripts, MLatom@XACS lowers the barrier to entry for complex molecular simulations. As we continue to enhance and integrate this ecosystem, we anticipate it will become an indispensable resource for theoretical chemists worldwide.
MB
Reference
[1] L. Zhang, S. V. Pios, M. Martyka, F. Ge, Y.-F. Hou, Y. Chen, L. Chen, J. Jankowska, M. Barbatti, P. O. Dral, MLatom Software Ecosystem for Surface Hopping Dynamics in Python with Quantum Mechanical and Machine Learning Methods, J. Chem. Theory Comput. (2024). DOI: 10.1021/acs.jctc.4c00468