
How well can nonadiabatic dynamics forecast an experiment?
In brief:
- The Journal of Chemical Physics proposed a double-blind prediction challenge to forecast the outcomes of an upcoming experiment on cyclobutanone.
- We report our results based on surface hopping using five different electronic structure methods.
- We (and the other groups answering the challenge) observed sticking differences between results, especially due to electronic structure methods.
The philosopher of science Imre Lakatos proposed that we should judge a research program as progressive if it predicted novel empirical facts and that at least some could be tested. If not, the program is degenerating. Excited-state molecular dynamics constitutes a research program in Lakatos’ sense of a core set of theoretical assumptions (the Born-Oppenheimer separability, for example) surrounded by a protective belt of auxiliary hypotheses (the need for nonadiabatic corrections).
In 2023, The Journal of Chemical Physics proposed a double-blind prediction challenge to forecast the outcomes of an upcoming experiment using ultrafast electron diffraction. A gas sample of cyclobutanone would be irradiated with a 200 nm light source targeting the Rydberg excited state (n→3s), and electron diffraction images would be collected in variable step sizes. Theoreticians were invited to publish their simulations in a special issue, where only then would the experimental results be revealed.
Fifteen groups, ours inclusive [1], answered the challenge in early 2024. They submitted excited-state molecular dynamics simulations based on quantum wave packet propagations, multiple spawning, multiple cloning, Ehrenfest, and diverse surface hopping approaches. These simulations employed electronic-state surfaces computed with many quantum-chemical methods ranging from TDDFT to CASPT2. The results were strikingly different, with the (n→3s) state lifetime varying over four orders of magnitude and divergent reaction channels.
Our results, based on the same surface hopping setup but with different electronic structures [MCSCF, CC2, ADC(2), TDDFT, and MRCI/ODM3], exemplify such a divergence. All approaches are well established, have low computational costs, and, except for MCSCF, are straightforward to use.
Each of these approaches was expected to offer specific advantages and technical difficulties. MCSCF and MRCI/ODM3 should suffer from instabilities caused by changes in the active space orbital composition during dynamics. MCSCF mostly neglects dynamic electron correlation. MRCI/ODM3 should poorly describe diffuse states, such as S2, in cyclobutanone. The single-reference character of TDDFT, CC2, and ADC(2) may cause trouble during dissociation. They are also not expected to work well near S1/S0 crossings. CC2, as a non-Hermitian method, should not converge when S2 and S1 are degenerated. ADC(2) is well known for incorrectly describing the excited state of carbonyl compounds.
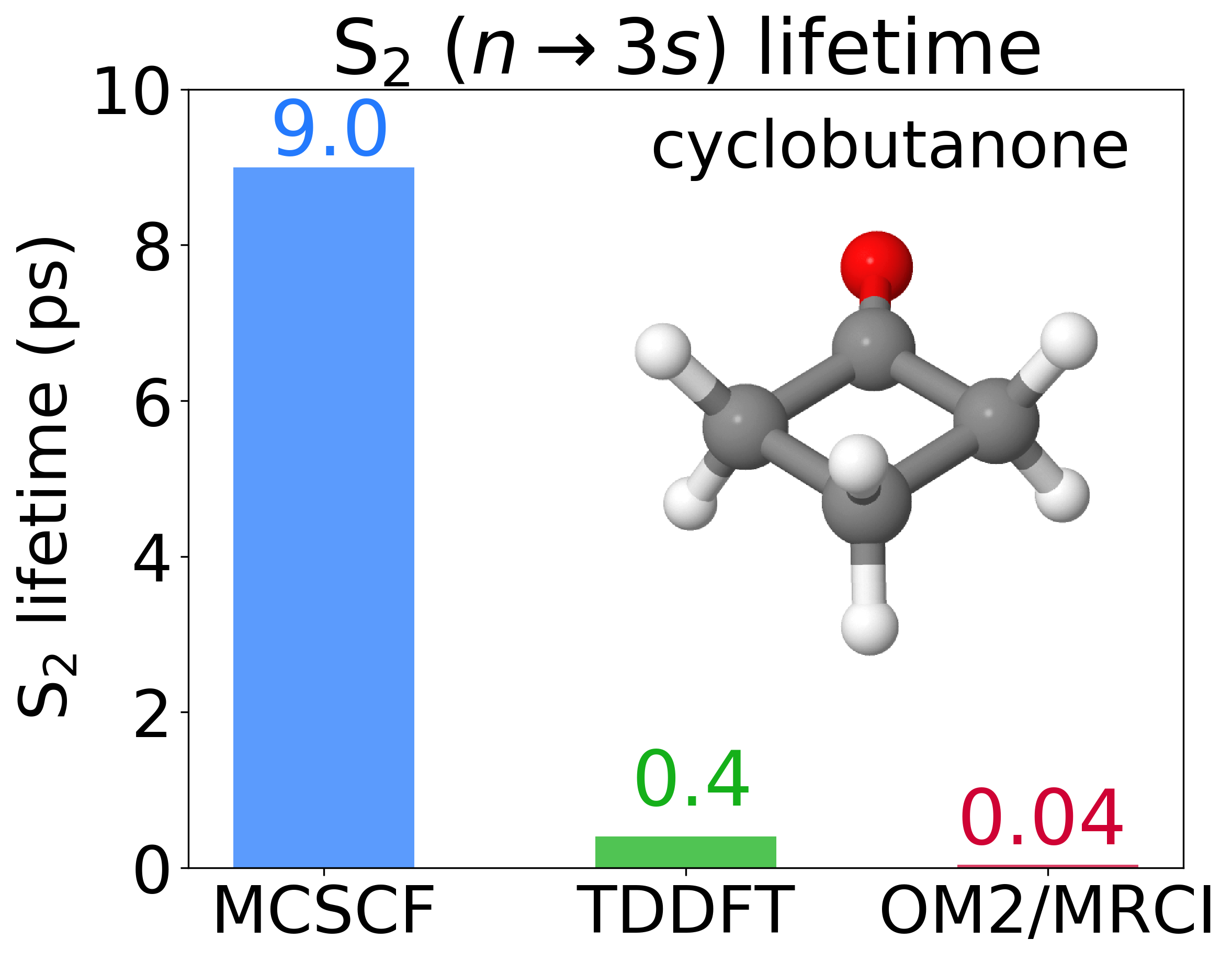
While MCSCF predicted a nine-ps lifetime, TDDFT was 0.4 ps, and MRCI/OM2 was 0.04 ps. ADC(2) predicted 0.9 ps, but its estimate must consider that we are dealing with carbonyl compounds. As expected, CC2 dynamics failed due to non-convergence issues. Our paper also includes a Table summarizing the results from all 15 papers in the Prediction Challenge.
On the other hand, MCSCF dynamics with either analytical nonadiabatic coupling vectors or approximated topography-based couplings (TD-BA) yielded similar results, even for large energy-gap hoppings, a situation particularly challenging for TD-BA.
Nonadiabatic dynamics simulations have proved invaluable for helping assign experimental features, rationalize empirical data, and explore the configurational space to reveal excited state reaction mechanisms far from our chemical intuition. Nevertheless, these conflicting dynamics results draw a worrisome picture of a field where routine nonadiabatic dynamics methods may have low prediction power.
We hope the debate on these divergent results will lead us to more accurate and precise methodologies, helping to unveil the best practices and development directions we must follow to guarantee that our research field remains progressive.
MB
Reference
[1] S. Mukherjee, R. S. Mattos, J. M. Toldo, H. Lischka, M. Barbatti, Prediction Challenge: Simulating Rydberg photoexcited cyclobutanone with surface hopping dynamics based on different electronic structure methods, J. Chem. Phys. 160, 154306 (2024). DOI: 10.1063/5.0203636