

This article offers a lifeline for those lost in the sea of molecular machine learning potentials.
In brief:
- We computed a benchmark of ground-state energies for several molecules computed with many machine learning potentials (MLP).
- The benchmark considers molecular sizes and conformations, simulation times, prediction accuracies, and training set sizes to deliver rules to tell the optimal MLP for each situation.
Quantum-chemistry simulations based on potential energy surfaces of molecules provide invaluable insight into the physicochemical processes at the atomistic level and yield such important observables as reaction rates and spectra. Machine learning potentials (MLP) promise to significantly reduce computational costs and enable otherwise unfeasible simulations. However, the surging number of such potentials begs the question of which one to choose or whether we still need to develop yet another one.
In a work led by Max Pinheiro Jr in collaboration with Pavlo Dral (Xiamen University), we address this question by evaluating the performance of popular machine learning potentials in terms of accuracy and computational cost.
Our first goal was to deliver structured information for non-specialists in machine learning to guide them through the maze of acronyms, recognize each potential’s main features, and judge what they could expect from each one.
For instance, when people say they used ANI or an sGDML model to compute the potential energies of a molecule, what does that precisely mean?
An MLP model is always composed of two elements, an ML algorithm and a descriptor. ANI, for instance, is a neural network algorithm with a fixed, local descriptor (or NN/LD-f to shorten). In turn, sGDML uses a kernel method as an algorithm and a global descriptor (KM/GD). Give it a goal. Look at the figure below and tell which kind of MLP would be PhysNet?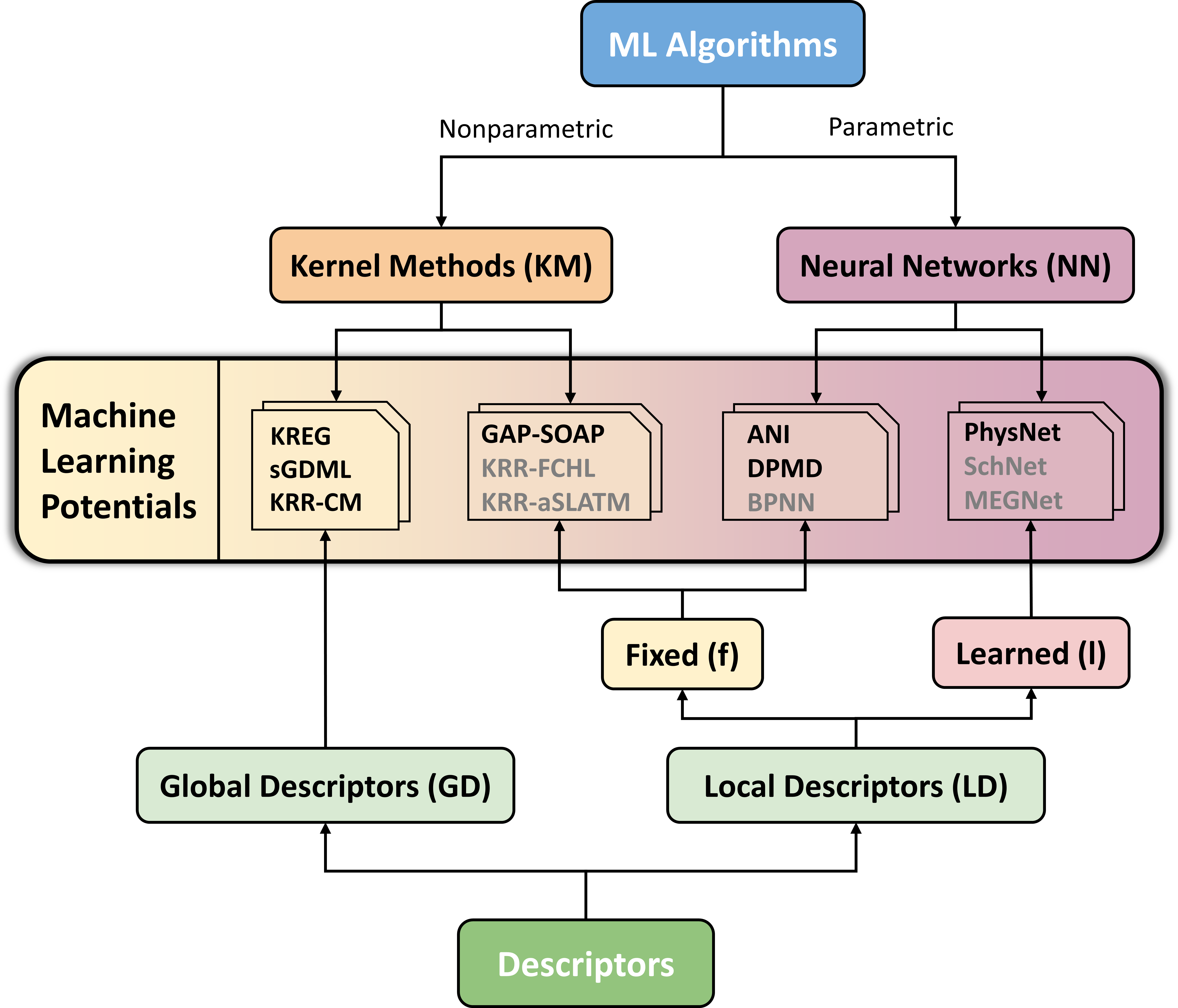
If you answered NN/LD-l, you’ve got the idea.
In the paper, we surveyed the main algorithms and descriptors. However, the paper’s core is the discussion of how some of the main MLPs currently available perform.
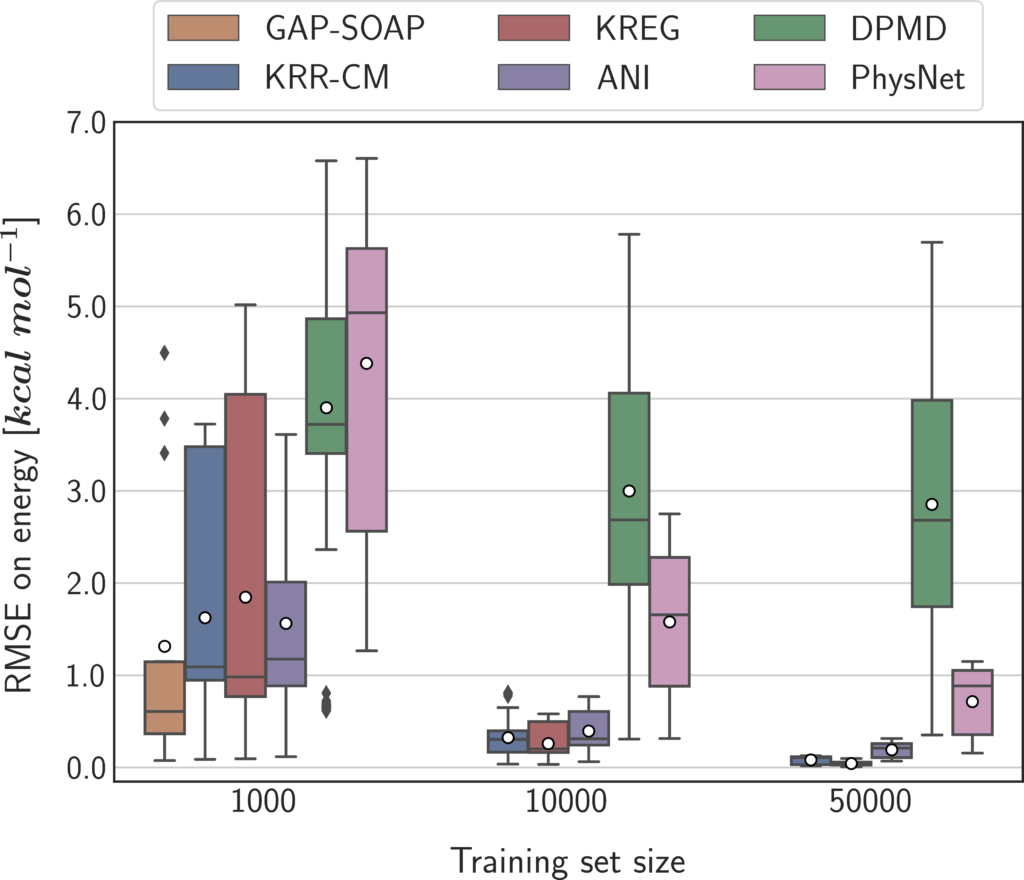
We built a benchmark considering molecular size, number of training set points, and availability of quantum-mechanical energy gradients, testing seven types of MLP models based on both NN and KM algorithms. I’m not going into the technicalities here, but the simulations were designed to compare them on an equal footing. Moreover, reproducibility was a serious concern. We adopted a unified open-source software interface (using MLatom), always used the same hardware, and made all data (raw and processed) available.
Our analysis delivered some general guidelines to choose the adequate MLP for each case, optimizing accuracy and computational times. These guidelines are summarized in the decision tree shown in the next figure.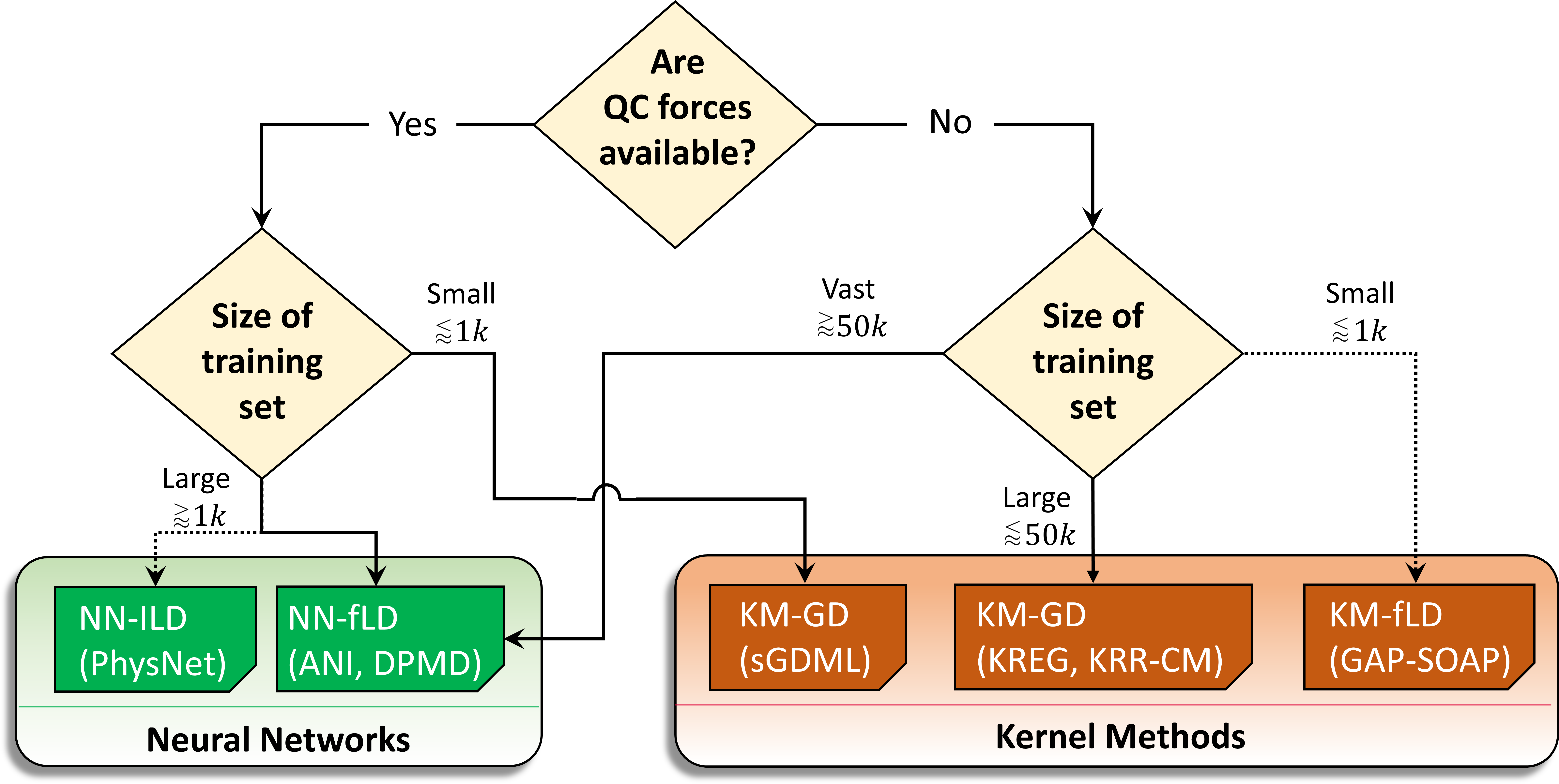
For instance, if quantum-mechanical energy gradients (forces) are available (like in TDDFT), and you are using a small training set (about 1000 points), the best option may be using a kernel model with a global descriptor, like in sGDML.
In general, if forces aren’t available (like in CASPT2), kernel methods are the best option unless you need vast training sets with over 50 thousand points. When forces are available, however, neural networks may be better.
This benchmark is just a static snapshot of a very dynamics field. However, we have provided a protocol to be followed in the future, to evaluate new MLPs. Our vision is to have an open-access platform, available on MLatom.com/MLPbenchmark1,
collecting up-to-date comparisons between different MLPs on equal grounds.
MB
Reference
[1] M. Pinheiro Jr, F. Ge, N. Ferré, P. O. Dral, and M. Barbatti. Choosing the right molecular machine learning potential. Chem. Sci., 2021, DOI: 10.1039/D1SC03564A.