

New dynamics method for dissipative processes.
In brief:
- There are many molecular processes in which the norm of the wave function isn’t conserved.
- They are usually modeled by complex-valued potential energy surfaces.
- We propose CS-FSSH, a surface hopping variant using complex PES.
Despite the continuous development of theoretical methodologies for describing the nonadiabatic dynamics of molecular systems, there is a lack of multidimensional approaches for processes where the norm of the wave function isn’t conserved, as when a set of discrete states interacts with a continuum of states.
This class of problems requires the use of non-Hermitian quantum mechanics, which can be modeled with imaginary potential terms. These terms mimic the decay from the discrete states and account for the decrease of the wave function norm. Current approaches rely on building complex-valued PES of reduced dimensionality, which is not optimal for more involving and realistic multidimensional problems.
Fabris and I present a novel methodology for describing the dynamics of dissipative systems. It is a generalization of the trajectory surface hopping (TSH) method to the case of complex-valued PESs, while employing the popular fewest switches surface hopping (FSSH) for the hopping probabilities. As such, we name it complex surface fewest switches surface hopping, or CS-FSSH for short.
The comparison of the CS-FSSH predictions with respect to quantum dynamics results for a one-dimensional three-states model is outstanding, as shown in the figure below. Both approaches provided quite similar behaviors for the populations, in two distinct couplings regimes.
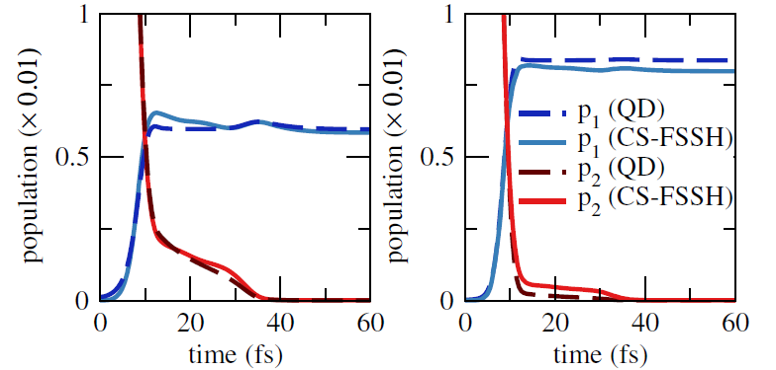
Comparison between exact quantum results and CS-FSSH predictions for a 1-D model with strong coupling (left) and weak coupling (right).
On-the-fly dynamics simulations of a vast class of processes can be envisioned with the CS-FSSH methodology, including:
- autoionization from transient anions,
- core-ionized and superexcited states,
- Auger and interatomic Coulombic decay,
- time-dependent luminescence,
- decoherence, …
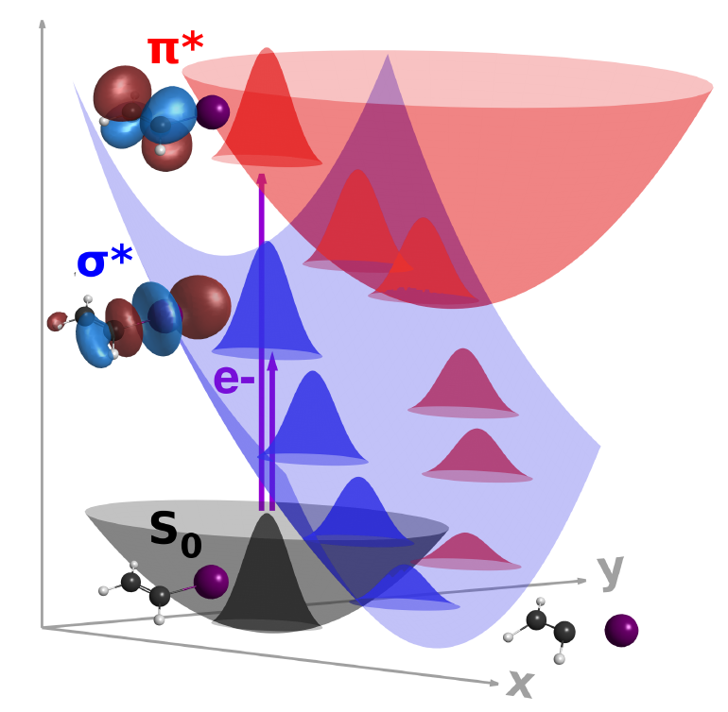
As a first application of the CS-FSSH methodology, we have investigated the low energy electron induced dynamics of iodoethene. The molecule is expected to display a prototypical π*/σ* indirect dissociative electron attachment (DEA) mechanism, which should also be encountered in biologically relevant halogen-containing molecules.
CS-FSSH showed that the electron capture into the π* orbital promotes C=C stretching and out-of-plane vibrations, followed by charge transfer from the double bond into the σ* orbital at the C-I bond, and, finally, the release of the iodine ion, all within only 15 fs. The CS-FSSH and experimental DEA cross-sections as a function of the electron energy are shown in the next figure.
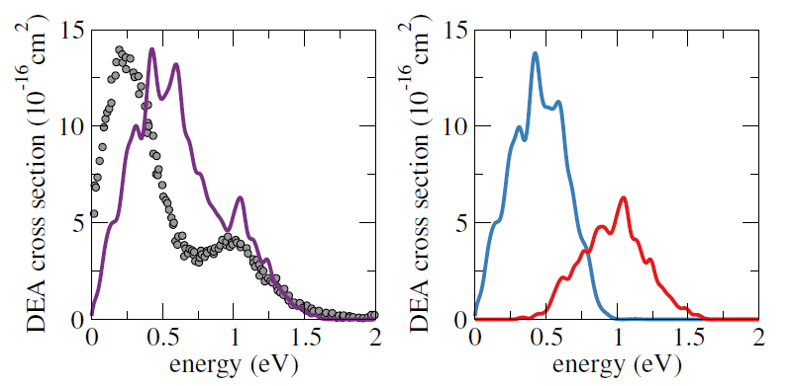
Calculated DEA (dissociative electron attachment) cross-section for iodoethene (purple), when starting from the lower (blue) or the upper (red) anion states, compared to available experimental data (grey dots).
While this π*/σ* mechanism is widely accepted to take place in halogenated species, being often evoked to interpret experimental data, it had never been theoretically demonstrated.
CS-FSSH has been implemented in a development version of Newton-X, and it will be available in the next release of the program.
MB
Reference
[1] F. Kossoski, M. Barbatti, Nonadiabatic Dynamics in Multidimensional Complex Potential Energy Surfaces, Chem. Sci. DOI: 10.1039/d0sc04197a (2020).