Dynamics reveals how to design chemical substitutions to control proton transfer efficiency.
Excited state intramolecular proton transfer (ESIPT)—an ultrafast photoinduced transformation in which a proton moves between two electronegative centers—is an exciting, interdisciplinary field of study, attracting interest of chemists, physicists, engineers, and biologists.
Acting at ultrashort timescales, with the transfer taking place within few tens of femtoseconds, ESIPT allows for a fast redistribution of excitation energy and, thus, enhances the photostability of the excited molecular system. The proton transfer also induces a pronounced redistribution of the electronic density, opening possibilities for applications in bioluminescence color tuning, selective photoswitching, and information storage.
In a project led by Joanna Jankowska (currently at the University of Southern California) and in collaboration with Joanna Sadlej and Andrzej Sobolewski, we have studied the photodeactivation process of an aromatic Schiff base model, salicylidene methylamine (SMA), and two of its derivatives, 6-cyano-salicylidene methylamine (6-CN-SMA) and 3-hydroxy-salicylidene methylamine (3-OH-SMA).
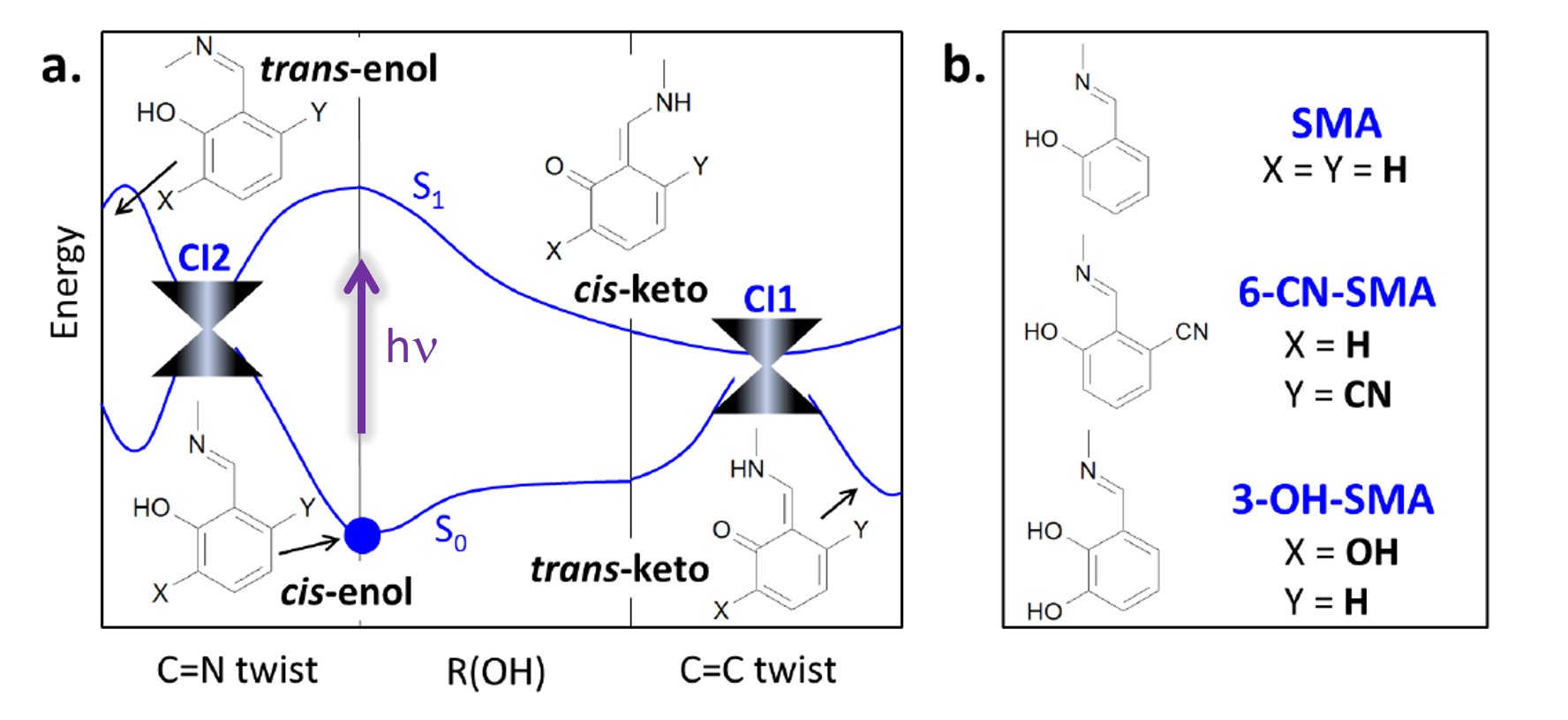
The general deactivation profile (see figure above) shows that there is two pathways in competition. After excitation, the molecules may either do a proton transfer and reach the conical intersection at the cis-keto conformation (CI1) or they may not do the proton transfer, and instead follow a C=N twist, reaching conical intersection CI2.
Naturally, the ratio between the two pathways will depend on the specific chemical structure. For instance, SMA has a high ESIPT efficiency: 92±6%, as predicted by our dynamics simulations. If we add a π-electron-acceptor group (-OH) to SMA (forming 3-OH-SMA), the ESIPT efficiency doesn’t change significantly: 86±8%. However, if instead we add a π-electron-donor group (-CN) to SMA (forming 6-CN-SMA), the ESIPT efficiency drops to 58±11%.
We show that these differences in the ESIPT efficiency is connected to the dominant character of the lowest excited singlet state—ππ* vs. nπ*. We also show that the relative energetic alignment of the ππ* and nπ* states may be controlled through chemical substitutions made to the aromatic ring of the Schiff-base molecule.
Thus, the structural modifications of the chromophore adding -OH or -CN groups open a way to control: (1) the ESIPT vs. C=N twist splitting ratio, (2) the accessibility of different conical intersections, and (3) barrierless vs. barrier-hindered ESIPT processes.
These findings will improve the rational design strategies employed for ESIPT systems, especially in the context of photoswitching.
Reference
J. Jankowska, M. Barbatti, J. Sadlej, and A. L. Sobolewski, Tailoring the Schiff Base Photoswitching – nonadiabatic molecular dynamics study of substituent effect on Excited State Proton Transfer; Phys. Chem. Chem. Phys., doi:10.1039/c6cp08545h (2017).