So far, the long lifetime of UV-excited thymine has been explained in terms of trapping in the second excited state. Simulations with electron correlation show this hypothesis doesn’t hold.
After UV excitation, gas phase thymine returns to the ground state in 5 ps, showing multiple time constants. This lifetime is significantly longer than those for the other isolated nucleobases, about 1 ps. There is no consensus on how to this process occurs in thymine, with a dispute between diverse models.
One of the first photophysical models to explain thymine’s deactivation was proposed by Domcke and co-workers, on the basis of analysis of ab initio potential energy surfaces. This model— we call it the “S1 trapping model”—assigns a sub-picosecond time constant to a fast S2(ππ*) → S1(nπ*) transition, while the 5 ps time constant is attributed to an S1(nπ*) → S0 transition. Thus, according to this interpretation, the elongated lifetime of thymine would be caused by a trapping in the S1 state.
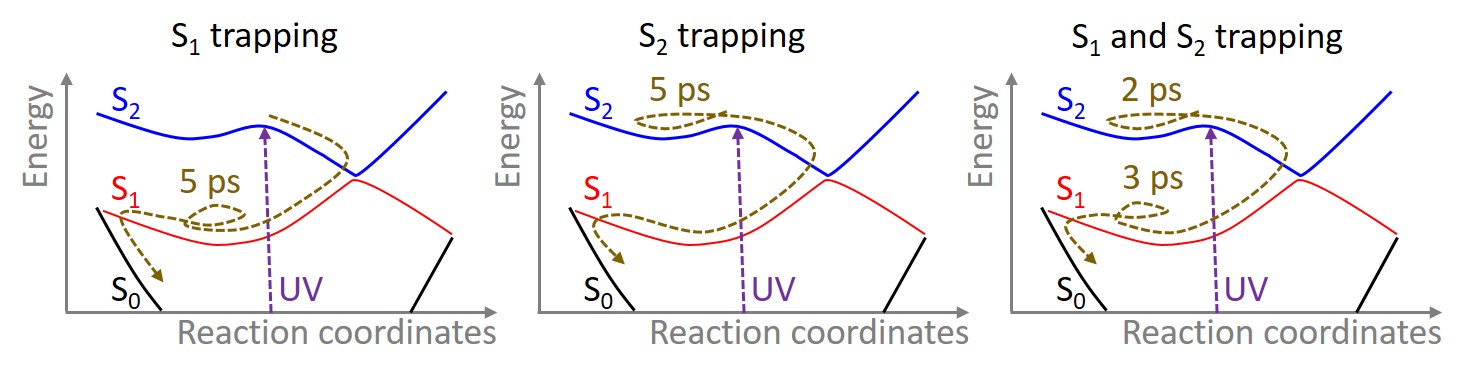
A different model for thymine’s deactivation was later proposed by Martinez ad co-workers, on the basis of multiple spawning dynamics on CASSCF surfaces. These simulations showed that, after excitation into S2(ππ*) state, conversion to S1(nπ*) was unexpectedly slow. This led to the hypothesis that thymine’s elongated lifetime was due to trapping in S2.
The “S2 trapping model” got some additional support from our surface hopping dynamics still on CASSCF surfaces. However, the surface hopping results also added a new layer of complexity, as they showed that the S2 trapping could only explain a delay of about 2 ps in the lifetime; therefore, to reach a 5 ps time constant, thymine should also be trapped in S1 after the S2 → S1 transition, pointing out to a new “S2 and S1 trapping model”.
A couple of years ago, the S2 trapping model was challenged by time-resolved Auger spectroscopy, which, combined with spectrum simulations at CIS level, made a good case towards a population transfer to S1(nπ*) state within 200-300 fs. Once more, the S1 trapping model was invoked to explain the picosecond time constant.
Giving this cloudy state of affairs, we decided to revisit thymine dynamics [1]. Although multiple spawning and surface hopping dynamics have provided some compelling arguments for the S2 trapping, these simulations have a common major weak point: they were based on CASSCF surfaces. CASSCF does an excellent job recovering non-dynamic electron correlation near intersections between the ground and the first excited states, however, it neglects most of dynamic electron correlation, which is present through the whole reaction path.
And this poses a serious problem: the key step to determine the occurrence (or not) of the S2 trapping is the S2 dynamics up to the S2/S1 crossing. In this region of the potential energy surface, we do not expect any relevant impact of non-dynamic electron correlation, but we are sure that dynamic electron correlation plays a role; for instance, correcting the strong overestimation of the ππ* energy typical of CASSCF predictions.
Therefore, we have approached the problem through surface hopping simulations based on ADC(2) method, which, quite opposite to CASSCF, recovers well dynamic correlation, but neglects non-dynamic correlation. This methodological change had a major impact on the results: the S2 trapping is strongly reduced, with 84% of the population converting to S1 in less than 1 ps.
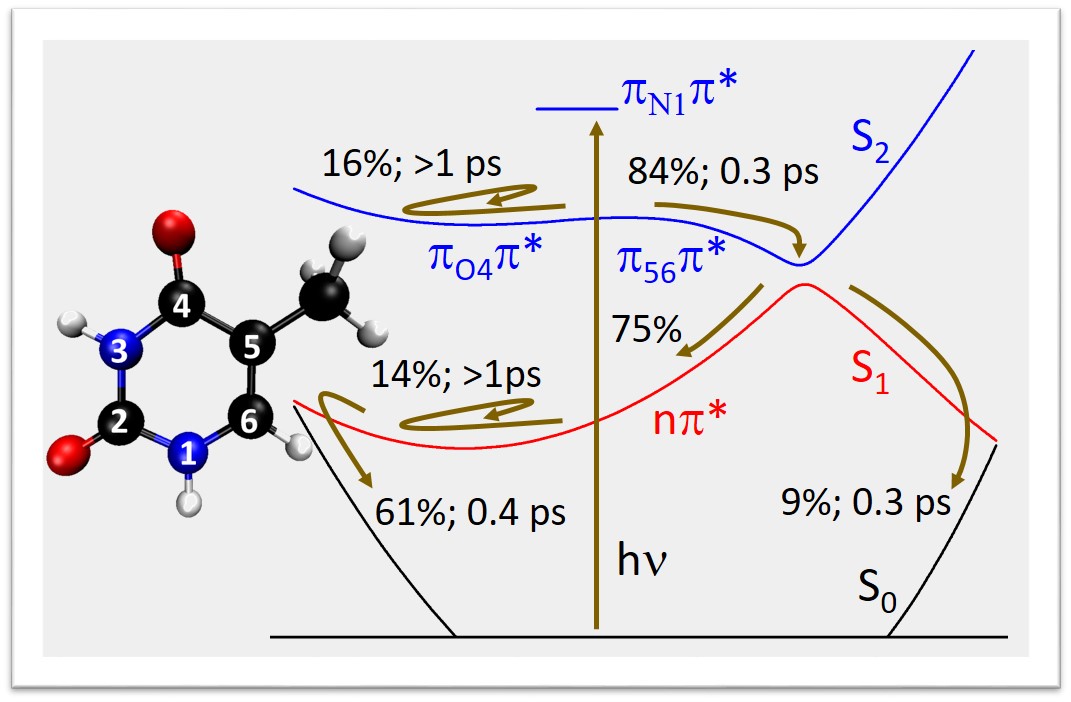
Because of the strong approximations in ADC(2), we do not believe that this result will be the last word on thymine’s dynamics. But at least it clearly shows that dynamic electron correlation plays a major role in the photophysics of this molecule and cannot be neglected.
These results were recently published in a special issue of Molecules on photochemistry of bioorganic molecules [1].
The paper also describes the implementation into Newton-X of the orbital derivative (OD) method developed by Artur Izmaylov’s group for fast computation of numerical nonadiabatic couplings.
Reference
[1] L. Stojanovic, S. Bai, J. Nagesh, A. F. Izmaylov, R. Crespo-Otero, H. Lischka, M. Barbatti, New Insights into the State Trapping of UV-Excited Thymine, Molecules 21, 1603 (2016). doi:10.3390/molecules21111603
3 Comments
A kinetic model for singlet oxygen photogeneration | Light and Molecules · October 12, 2017 at 8:16 AM
[…] Revisiting the excited-state dynamics of thymine […]
Singlet oxygen: rates strongly depend on geometry | Light and Molecules · October 26, 2017 at 7:28 AM
[…] Revisiting the excited-state dynamics of thymine […]
Dynamics of UV-Excited Thymine-Water Cluster – Light and Molecules · August 12, 2018 at 2:53 PM
[…] isolated thymine in the gas phase is excited by UV radiation, it quickly gets rid of the photoenergy by transforming it into […]
Comments are closed.